 Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 5:55 PM
|
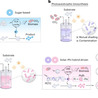
Solar-driven biosynthesis from carbon dioxide can facilitate sustainable chemical manufacturing. However, cell-density limits and contamination susceptibility have seriously hampered its practical implementation. Here we develop a solar–chemical hybrid-driven biosynthesis (SCHB) strategy integrating wastewater-based phosphorus recovery into biological photosynthetic metabolism for chemical production. Specifically, we incorporate the phosphite oxidation pathway into cyanobacteria to supplement additional electrons to stimulate bacterial growth. This strategy conferred contamination resistance and enabled the utilization of phosphite-rich wastewater. A series of chemicals including raspberry ketone, indigo and its derivatives were effectively synthesized via SCHB, and the synthesis efficiency was promoted by a factor of up to 305%. Furthermore, the scalability of SCHB was demonstrated at the 500-litre level with real wastewater, which synchronized chemical production with nutrient recovery. Life-cycle assessment and techno-economic analysis indicated notable environmental benefits and economic feasibility. This study potentially opens a viable approach for the sustainable biosynthesis of chemicals. Bio-photosynthesis has the potential to achieve sustainable chemical production, but technical challenges remain. This work proposes a solar–chemical hybrid-driven biosynthesis strategy to achieve efficient chemical production from wastewater and carbon dioxide.
|
|
Scooped by
mhryu@live.com
Today, 5:47 PM
|
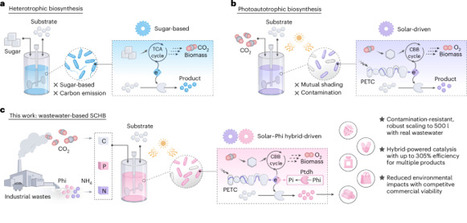
Microbial cell factories offer a sustainable route to plant-derived natural products, but yield drift and strain degeneration persist. Growth-coupled biosynthesis can continuously enrich high producers, yet specific product-responsive biosensors remain scarce and their population-level effects are unclear. Here, we describe a rapid transcriptome-mining workflow that, as proof-of-concept, delivers yeast biosensors for glycyrrhetinic acid and medicarpin. By fine-tuning PDR5 promoter, we expand the dynamic range of the glycyrrhetinic acid sensor and wire it to an essential gene, establishing a growth-addiction circuit that increases titer by 46.8 % after subculture. Single-cell transcriptome reveals that the evolved strain population exhibits a completely different division of labor compared to the initial strain. Coupling does not eliminate phenotypic heterogeneity; instead, it amplifies a dedicated sub-population marked by discrete transcriptional signatures. Deletion of genes highly expressed in non-producing cells or enrichment of high-producing cell clusters can further boost population-level production. This study provides both a generalizable biosensor-discovery platform and single-cell-guided strategies for stabilizing and optimizing natural-product cell factories. Growth-coupled biosynthesis in microbes can stabilise production yields, yet their population-level effects are unclear. Here the authors use ScRNA-seq to reveal that an evolved glycyrrhetinic acid-producing growth-coupled yeast strain population achieves division of labor.
|
|
Scooped by
mhryu@live.com
Today, 3:56 PM
|
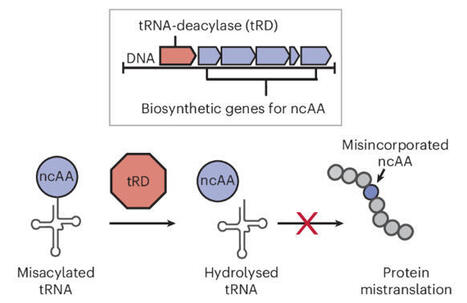
Amino acids are one of nature’s most privileged building blocks for generating molecular diversity on length scales ranging from small molecules to proteins. While amino acid products arising from certain class-defined biosynthetic pathways can be found with established bioinformatic strategies, those that fall outside of these types remain difficult to identify. Here, to address this challenge, we have developed an approach to find biosynthetic gene clusters (BGCs) that utilize and modify amino acid monomers while remaining agnostic to biosynthetic class. We demonstrate that tRNA deacylases specific for host-synthesized non-canonical amino acids (ncAAs) serve as a common genomic marker of ncAA metabolism. Using this approach, we show that thousands of cryptic BGCs can be identified and demonstrate the discovery of BGCs for several distinct ncAAs as well as a hydrazide-containing tripeptide. We anticipate this approach will have broad applications for discovering natural products with ncAAs and beyond. tRNA deacylases have evolved as resistance genes towards natural products that contain non-canonical amino acids by preventing their mistranslation. Now a strategy has been developed that leverages tRNA deacylases as class-agnostic genomic markers for amino acid-based biosynthetic gene clusters, identifying thousands of cryptic clusters and enabling the discovery of amino acid-based natural products.
|
|
Scooped by
mhryu@live.com
Today, 3:39 PM
|
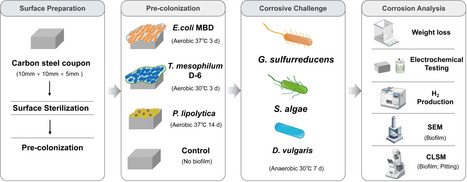
Sustainable materials are needed to address the serious economic and safety risks of microbial metal corrosion. Colonizing metal surfaces with biofilms of noncorrosive microbes was previously shown to reduce aerobic, abiotic corrosion. However, the ability of biofilms to thwart highly corrosive anaerobic microbes is untested. Here we report on a strain of Escherichia coli genetically modified for enhanced metal adherence and adaptively evolved to tolerate sulfide. The E. coli biofilms effectively inhibited all known major routes for anaerobic microbial iron corrosion, including proton and sulfide attack, as well as the highly aggressive corrosion of electroactive microbes that directly extract electrons from Fe0. The E. coli biofilms prevented corrosion much better than biofilms of other microorganisms previously reported to reduce aerobic, abiotic corrosion. The results highlight the possibility of tailoring biofilm properties to function as effective sustainable, self-healing coatings to safeguard critical metal infrastructure.
|
|
Scooped by
mhryu@live.com
April 12, 11:58 PM
|
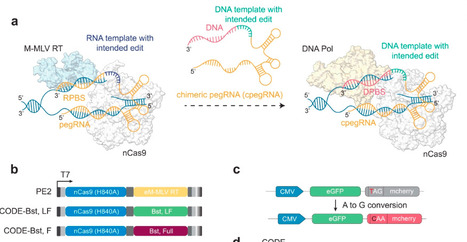
Prime editing has emerged as a precise and powerful genome editing tool, offering a favorable gene editing profile compared to other Cas9-based approaches. Here we report several nCas9-DNA polymerase fusion proteins and their engineered versions to create a simple and efficient two-component chimeric oligonucleotide-directed editing (CODE) system. CODE contains a derivative of Bst DNA polymerase engineered for increased thermostability and processivity as well as a chimeric pegRNA (cpegRNA) for programmable search and replace genome editing. Additionally, CODEMax(exo+) features a 5’ to 3’ exonuclease activity that promotes effective strand invasion and repair outcomes favoring the incorporation of the desired edit. We demonstrate that CODEs can perform small insertions, deletions, and substitutions with improved efficiency compared to PEMax at many loci in HEK293T cells with plasmid- and RNP-based delivery. We also show that CODEMax can successfully modify mouse and bovine embryos with up to 9.3% precise editing. Further optimization of CODEMax systems may enhance editing outcomes in embryos and other challenging contexts. Overall, CODEs complement existing prime editors to expand the toolbox for genome manipulations without double-stranded breaks. Gene editing technologies enable precise DNA modifications but remain limited by efficiency, precision of intended edits, and flexibility. Here, authors develop chimeric oligonucleotide-directed and Cas polymerase-based systems that enhance programmable gene correction across cells and embryos.
|
|
Scooped by
mhryu@live.com
April 12, 9:31 AM
|
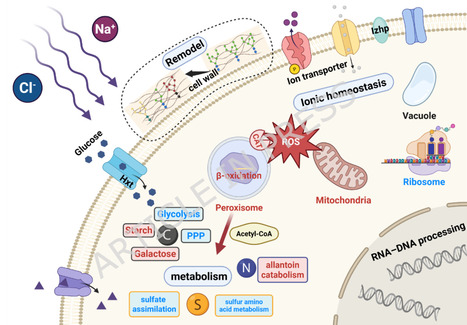
High-salinity conditions frequently impair the fermentation performance of Saccharomyces cerevisiae in industrial processes involving high-osmolarity substrates. Identifying genetic determinants that enhance salt tolerance is therefore essential for the development of robust yeast cell factories. In this study, a comparative transcriptomic analysis was performed to investigate the transcriptional responses of a salt-tolerant strain, E-158, and its parental strain, KF-7, under 1.25 M NaCl stress, with the aim of identifying potential targets for strain engineering. Comparative transcriptomic analysis revealed extensive transcriptional differences between E-158 and KF-7 under high-salt conditions, involving central carbon and nitrogen metabolism, peroxisome-associated oxidative stress responses, ion transport, cell wall-related processes, and sporulation-related pathways. Based on these profiles, two transcription factors (CUP9 and ZNF1) and three functional genes (DAL1, IDP2, and CTA1) were selected for functional validation. Overexpression or deletion of the transcription factors, as well as overexpression of the functional genes, was carried out in KF-7. Fermentation experiments under 1.25 M NaCl demonstrated that all engineered strains outperformed the parental strain. Among them, overexpression of CTA1 resulted in the greatest improvement, with glucose consumption and ethanol production increased by 35.04% and 45.66%, respectively, after 96 h of fermentation. This study demonstrates that comparative transcriptomics can serve as an effective strategy for identifying engineering targets associated with salt tolerance in S. cerevisiae. The evaluated genes provide potential targets for strain improvement and offer insights for the rational design of yeast cell factories suited for high-salinity biofuel and bioproduct fermentation processes.
|
|
Scooped by
mhryu@live.com
April 12, 9:14 AM
|
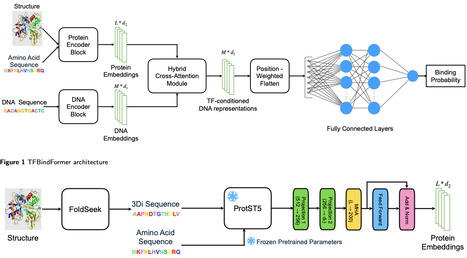
Transcription factors (TFs) are central regulators of gene expression, and their selective recognition of genomic DNA underlies various biological processes. Experimental profiling of TF -- DNA interactions using chromatin immunoprecipitation followed by sequencing (ChIP-seq) provides high resolution maps of in vivo TF -- DNA binding but remains costly, labor-intensive, and inherently low-throughput, limiting their scalability across different transcription factors,cell types, and regulatory conditions. Computational modeling therefore plays an essential role in inferring TF -- DNA interactions at genome scale. However, most existing computational models rely solely on DNA sequence and chromatin features to predict TF -- DNA binding, neglecting TF-specific protein information. This omission limits their ability to capture protein-dependent binding specificity. Here, we present TFBindFormer, a hybrid cross-attention transformer that explicitly integrates genomic DNA features with TF specific representations derived from protein sequences and structures. By modeling protein-conditioned, position-specific TF -- DNA interactions, TFBindFormer enables direct learning of molecular determinants underlying DNA recognition. Evaluated across hundreds of cell-type-specific TFs and hundreds of millions of genome-wide DNA bins, TFBindFormer consistently outperforms DNA-only baselines, achieving substantial gains in both area under precision-recall curve(AUPRC) and area under receiver operating characteristic curve(AUROC). Together, these results demonstrate that integrating TF and DNA features via cross-attention enables TFBindFormer to serve as an effective and scalable framework for large-scale TF -- DNA binding prediction.
|
|
Scooped by
mhryu@live.com
April 12, 12:17 AM
|
Intrinsically disordered protein regions are ubiquitous across all kingdoms of life. These structurally heterogeneous regions play central roles in cellular processes such as transcriptional regulation, cellular signaling, and subcellular organization, yet they have remained largely inaccessible to rational design. Structure-based generative methods are not applicable to proteins that lack a stable fold, and existing sequence-based approaches for disordered regions rely on sampling methods that do not capture the evolutionary statistics of natural disordered regions. Here, we introduce IDiom, an autoregressive protein language model trained on 37 million intrinsically disordered region sequences curated from the AlphaFold Database. Trained using a fill-in-the-middle data augmentation, IDiom generates disordered region sequences conditioned on their surrounding structured context, as well as fully disordered proteins without any context. The model generates diverse sequences that recapitulate biologically relevant sequence features of natural disordered regions, and we demonstrate that post-training via reinforcement learning with a subcellular localization reward model produces sequences with features which are consistent with known sequence determinants of compartment-specific localization. These results establish IDiom as a general platform for the generative design of intrinsically disordered proteins and regions.
|
|
Scooped by
mhryu@live.com
April 11, 11:31 PM
|
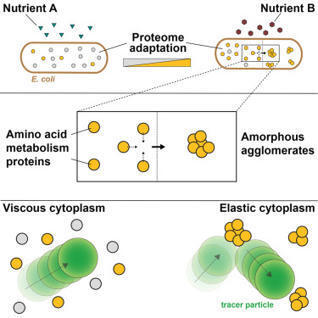
Molecular crowding in the bacterial cytoplasm restricts diffusion of large molecules, impacting cellular processes. To monitor cytoplasmic diffusion and rheology, we used single-particle tracking in E. coli, finding a 3-fold variation in the diffusion of a 40-nm particle across exponential growth conditions. Known determinants of rheology did not account for this variation. Instead, we found a strong anti-correlation between the diffusion coefficient and the abundance of amino acid metabolism proteins (clusters of orthologous groups [COG] category “E”), persisting upon genetic perturbations, and that lower diffusion is associated with increased elasticity. Photoactivated light microscopy revealed that some amino acid metabolism proteins form clusters. Electron microscopy showed that these proteins can form amorphous agglomerates at physiological concentrations in vitro due to their high hydropathy, which also confers low disorder and compactness. These findings show that diffusion is controlled by the formation of protein agglomerates and thus reveal how condition-induced proteome changes affect cytoplasmic rheology.
|
|
Scooped by
mhryu@live.com
April 11, 11:11 PM
|
The global rise of antimicrobial resistance (AMR) demands urgent attention. While genetic drivers are well studied, epigenetic mechanisms, particularly DNA methylation, are emerging as key contributors to bacterial adaptation under antibiotic pressure. This review examines the roles of N6-methyladenine (m6A), N4-methylcytosine (m4C), and 5-methylcytosine (m5C), each catalyzed by distinct DNA methyltransferases (MTases), in regulating resistance-related processes such as efflux pump expression, β-lactamase activity, and stress responses. Advances in long-read sequencing technologies, including SMRT and ONT, now enable single-base resolution detection of methylation and support strain-specific methylome mapping. These efforts reveal methylation patterns that are dynamic, strain-dependent, and environmentally responsive, complicating resistance profiling. Emerging applications for tackling methylation-linked AMR include methylation-aware diagnostics and CRISPR-based epigenetic editing. Tools like CRISPR-dCas9 fused to DNA methyltransferases enable targeted, reversible suppression of resistance genes regulated by methylation. Current findings position DNA methylation as both a regulator of AMR and a promising target for next-generation diagnostics and therapeutics. However, challenges remain, including the lack of validated biomarkers, inconsistent protocols, and difficulty interpreting mixed-species data. Integrating methylation profiles with transcriptomic and phenotypic data will be essential to fully understand and target resistance mechanisms.
|
|
Scooped by
mhryu@live.com
April 11, 5:09 PM
|
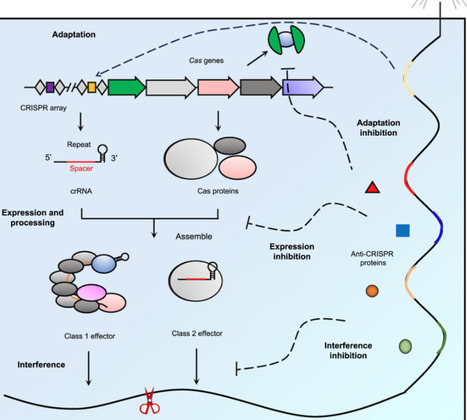
The CRISPR-Cas system constitutes an adaptive immune mechanism in prokaryotes that defends against mobile genetic elements. Within the perpetual co-evolutionary arms race between bacteria and their viral predators, bacteriophages encode anti-CRISPR (Acr) proteins that use sophisticated molecular strategies to sabotage CRISPR-Cas function. While canonical Acr proteins rely on steric blockade of Cas effectors, recent discoveries reveal unprecedented noncanonical mechanisms spanning CRISPR immunity stages. This review synthesizes recent mechanistic advances in this field since 2023, highlighting the expansion of noncanonical inhibition mechanisms beyond type I to include types II, V, and VI, as well as novel Acr interventions targeting multiple functional stages, such as spacer acquisition, translation-coupled inhibition, complex assembly/disassembly, and R-loop DNA binding. Structural insights demonstrate how Acr proteins achieve substoichiometric inhibition via conformational hijacking, catalytic repurposing, and molecular mimicry. Forged by the intense selective pressure of the phage–host conflict, these molecular innovations represent both remarkable evolutionary adaptations and versatile precision tools. They enable spatiotemporal control of CRISPR technologies, from engineered off-switches to diagnostic reset mechanisms, while posing critical challenges for therapeutic safety and microbiome management.
|
|
Scooped by
mhryu@live.com
April 11, 5:00 PM
|
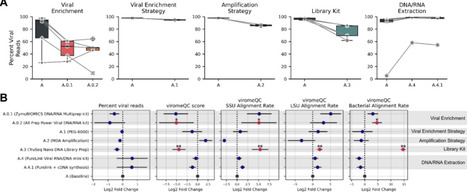
Human virome research is gaining increasing attention as viruses are recognized as critical modulators of microbial communities and human health. Viral metagenomics, however, faces unique challenges, including the low abundance and diversity of viruses in biological samples, the absence of universal marker genes, and biases introduced by experimental protocols. While various virome protocols have been benchmarked using viral particles or nucleic acids from mock communities, these approaches often fail to capture the complexity and heterogeneity of natural viromes. In this study, we systematically evaluated modifications to key methodological steps in the metagenomic analysis of human fecal samples, including viral enrichment, nucleic acid extraction, genome amplification, and library preparation. Using gold-standard bioinformatic approaches on sequencing datasets generated after amplification, we assessed the impact of these modifications on relative viral taxonomic assignment, contig quality, richness, diversity, and inferred genome structure. Our findings reveal striking trade-offs between recovery of viral genomes and retention of non-viral sequences, demonstrating how methodological choices can shape the inferred virome composition. Based on these observations, we propose an optimized protocol that enhances viral genome recovery while reducing contamination from non-viral sequences. This refined workflow provides a more robust and reliable framework for gut virome studies, paving the way for a deeper exploration of the role of viruses in human health and microbial ecosystems.
|
|
Scooped by
mhryu@live.com
April 11, 4:41 PM
|
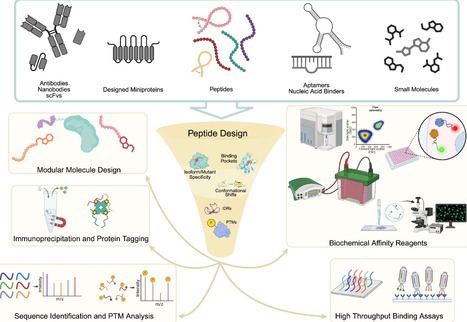
Peptides occupy a unique niche as biochemical tools: they are small, modular reagents capable of perturbing protein function with a precision that is often inaccessible to small molecules or antibodies. Historically, their broader use in biochemical research has been constrained by slow discovery workflows, limited control over specificity, and poor physicochemical properties. Recent advances in artificial intelligence have begun to change this landscape by enabling the rational, data-driven design of peptides tailored for specific experimental tasks. In this review, we focus on AI-designed peptides as practical tools for biochemistry. We survey sequence-based and structure-based design paradigms, highlighting how each supports distinct classes of peptide tools, including isoform- and motif-specific binders, multi-objective assay-ready reagents, and functional peptides that enable degradation, stabilization, or biophysical interrogation of proteins. By emphasizing experimental utility, design constraints, and appropriate use cases, we aim to provide a framework for selecting and deploying AI-designed peptides as on-demand reagents in modern biochemical research.
|
|
|
Scooped by
mhryu@live.com
Today, 5:55 PM
|
The rapid growth of genomic data and increasing adoption of long-read sequencing technologies have rendered variant calling one of the most computationally demanding tasks in genomic analysis. Although deep learning-based methods currently outperform conventional approaches in distinguishing true variants from complex sequencing noise, they impose prohibitive computational and time requirements. To address this limitation, we present a computational framework based on Clair3 that integrates parallelized feature generation, enhanced variant phasing, in-memory read haplotagging, and GPU-accelerated neural network inference to accelerate variant calling. By dynamically optimizing the use of both GPU and CPU resources, our method achieves substantial runtime improvements without compromising accuracy. We evaluated our framework across a range of sequencing depths, diverse samples, and multiple hardware configurations. Our results demonstrate that the optimized pipeline completes variant calling for a 30× whole-genome sequence in 12–20 minutes using standard computational resources (32 CPU threads and one NVIDIA GPU), and in 12–15 minutes on an Apple Mac Studio (32 threads), which is ∼10–20-fold speedup compared with its initial release. In addition to exceptional efficiency, our method maintains state-of-the-art accuracy, achieving SNP F1-scores of 99.32% and 99.70% on 30× ONT and PacBio GIAB HG003 datasets, respectively. This work introduces a rapid, accurate, and scalable variant calling framework that effectively supports large-cohort genomic studies and time-sensitive clinical applications.
|
|
Scooped by
mhryu@live.com
Today, 5:40 PM
|
From aircraft to neural networks, engineers are often inspired by biological systems; however, literature often conflates inspiration with scientific evidence. Here, I propose a transparent and consistent reporting framework for bio-inspired design that can accelerate technological progress and stimulate collaborative scientific pursuits. From aircraft to neural networks, engineers are often inspired by biological systems; however, literature often conflates inspiration with scientific evidence. Christina Harvey proposes a transparent and consistent reporting framework for bio-inspired design that can accelerate technological progress and stimulate collaborative scientific pursuits.
|
|
Scooped by
mhryu@live.com
Today, 3:51 PM
|
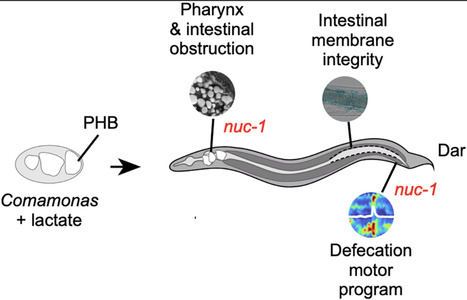
Bacteria, both individually and as symbionts of other organisms, significantly influence ecosystems by providing nutrients and metabolizing exogenous compounds. Some bacteria polymerize small organic acids such as lactate, pyruvate, and β-or 3-hydroxybutyrate when there is an excess of carbon relative to other elements. One such polymer, poly-β-hydroxybutyrate (PHB) is a biodegradable bioplastic. While the role of PHB as energy/carbon-storage in bacteria is well documented, the effects of PHB on interactions between bacteria and their hosts remain unclear. Here, we discover that PHB-producing bacteria can kill the nematode Caenorhabditis elegans. Death results from a combination of pharyngeal deformation, intestinal distention, disruption of the intestinal barrier, and defecation defects. Remarkably, mutations in C. elegans nuc-1, which encodes DNAse II, partially alleviate PHB-induced lethality. Altogether, our findings illustrate that PHB-producing bacteria can affect host-physiology and survival.
|
|
Scooped by
mhryu@live.com
Today, 12:23 AM
|
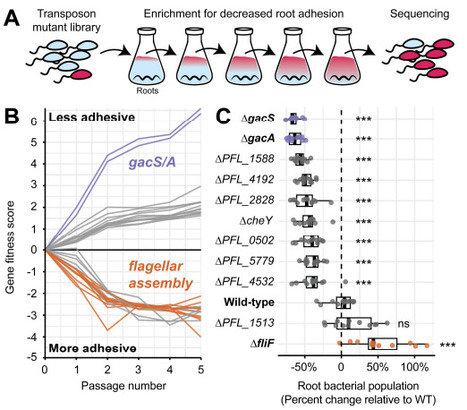
Plants host complex communities of microbes that are attached to root surfaces. While many studies have sampled mutant populations after prolonged incubation on roots to identify bacterial genes that enable long-term colonization, the molecular mechanisms governing the early stages root attachment remain less understood. Here, we developed an in vitro root culture system that enables controlled and scalable investigation of bacterial attachment to root tissue. We used this platform to perform a genome-wide screen for root attachment determinants in the plant-associated bacterium Pseudomonas protegens Pf-5. Our results reveal that the gacSA two-component system functions as a sensory integration hub for coordinating early root attachment. Mutations that disrupt gacS or gacA cause severe root attachment defects despite having no effect on abiotic surface attachment in standard biofilm assays. Mutation of flagellar assembly genes enhances root attachment by mimicking surface contact and activating the gac system. In parallel, chemical cues released by roots stimulate surface attachment in a gac dependent manner. By integrating these signals, the gac system activates cyclic di-GMP-mediated attachment programs that drive the transition from planktonic to sessile behavior required for root association. We build on this model to show that manipulating flagellar surface sensing enhances the competitive fitness of Pf-5 in the presence of a synthetic bacterial community, suggesting a strategy to improve the competitive fitness of beneficial microbes on crops. These findings establish a mechanistic framework linking surface sensing, global regulation, and root attachment in a beneficial rhizobacterium.
|
|
Scooped by
mhryu@live.com
April 12, 11:17 PM
|
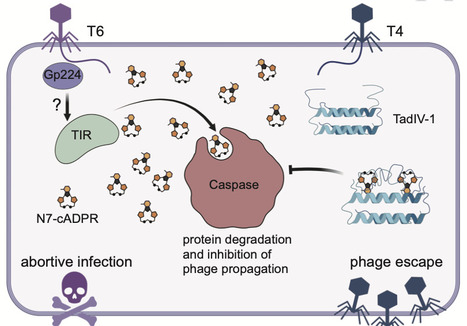
Triggering immune response through generation of signal molecules is a common immune strategy across all domains of life. In the bacterial type IV Thoeris antiphage system, a Toll/interleukin-1 receptor (TIR)-domain protein produces adenosine 5’-diphosphate-cyclo[N7:1”]-ribose (N7-cADPR) as the immune signal to activate a Caspase-like effector. Here, we identified an inhibitor of type IV Thoeris through a phage mating assay that allows a sensitive phage to acquire anti-defense genes from related resistant phages. The inhibitor (hereafter TadIV-1) functions as a sponge that sequesters the N7-cADPR signal to inhibit Caspase activation. Structural analyses of TadIV-1 indicate a distinctive signal binding mechanism, wherein the binding pocket comprises its N-terminal flexible loop. In addition, phages lacking TadIV-1 can escape type IV Thoeris sensing through mutation in the capsid vertex protein. Collectively, this work expands the phage anti-defense arsenal with a unique immune signal sequestration mechanism and provides insights into phage invasion recognition mechanism of type IV Thoeris. The bacterial type-IV Thoeris system produces, in response to phage infection, a signal molecule that triggers protein degradation and stops viral replication. Here, the authors identify a phage protein that sequesters the signal, thus blocking the antiviral response.
|
|
Scooped by
mhryu@live.com
April 12, 9:25 AM
|
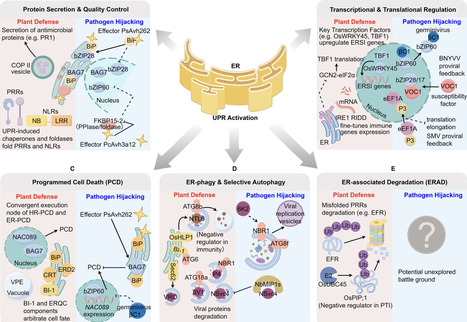
The endoplasmic reticulum (ER) stress response, a conserved proteostasis network, has emerged as a central hub that reprograms plant immunity during pathogen attack. This review synthesises how plants harness ER-stress signalling to mount multilayered defences and how pathogens have evolved counterstrategies to subvert these pathways. We delineate the molecular integration of the unfolded protein response (UPR) with canonical immune layers including pattern-triggered immunity (PTI), effector-triggered immunity (ETI) and systemic defences, highlighting salicylic acid (SA) and jasmonic acid (JA) as rheostats that fine-tune ER stress-immune crosstalk. Functionally, the UPR bolsters immunity by coordinating protein folding and secretion, reprogramming transcription and translation, activating ER-dependent programmed cell death (ER-PCD), and orchestrating ER-associated degradation (ERAD) and selective autophagy. Pathogens such as bacteria, oomycetes and viruses in turn deploy virulence factors that target UPR sensors and transcription factors, thereby attenuating ER-driven immunity. We propose a conceptual framework in which the outcome of UPR activation—resistance versus susceptibility—is determined by pathogen lifestyle, ER stress dynamics, subcellular compartmentalisation and pathogen effector intervention. We also consider biotechnological contexts in which strong transgene expression can itself provoke the UPR, and outline diagnostic experimental strategies to distinguish UPR-mediated effects from intended transgene functions. By integrating molecular mechanisms with pathogen counterstrategies, this review underscores the dynamic interplay between ER stress and immune signalling in plants and highlights opportunities to enhance crop resilience under global climate challenges.
|
|
Scooped by
mhryu@live.com
April 12, 12:25 AM
|
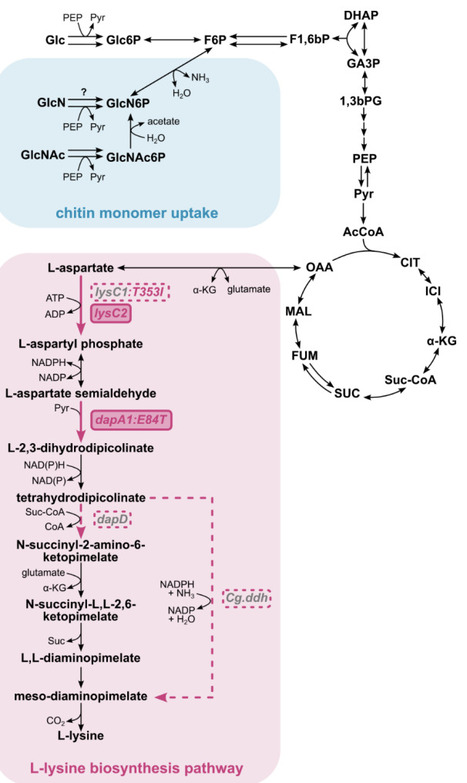
Despite its industrial importance, microbial L-lysine production has largely been confined to classical producer strains, leaving the fast-growing, non-pathogenic marine microorganism V. natriegens largely untapped as an unconventional biosynthetic platform. In this work, we established an L-lysine-overproducing V. natriegens DSM759 strain through a step-wise, systematic rational engineering strategy targeting the native biosynthetic pathway. Guided by our prior systems-level analysis of the strain′s genetic and regulatory architecture, we identified key metabolic bottlenecks and implemented knowledge-driven interventions to relieve pathway constraints. Central to production was alleviation of feedback inhibition in the native key regulatory enzymes, aspartate kinase (AK, lysC) and dihydrodipicolinate synthase (DHDPS, dapA). Site-directed amino-acid substitutions, replicating established E. coli feedback-resistance mechanisms, were introduced into conserved regions of the V. natriegens DSM759 enzymes, producing L-lysine-insensitive variants with kinetic parameters comparable to that of corresponding wild type enzymes. Among the tested configurations, the strain co-expressing Vn.lysC2 and Vn.dapA1:E84T reached the highest L-lysine titer (9.0±0.6 mM) and yield (0.11±0.01 molLys molGlc-1), whereas overexpression of additional L-lysine pathway genes provided no further benefit. Leveraging the host′s metabolic versatility, L-lysine synthesis was also demonstrated from the chitin-derived amino-sugar N-acetylglucosamine (0.09±0.00 molLys molGlcNAc-1), highlighting the potential to valorize chitin-rich waste streams from the seafood industry. This work establishes a minimal, rational strategy for L-lysine biosynthesis in V. natriegens DSM759 and positions it as a promising platform for sustainable amino acid production.
|
|
Scooped by
mhryu@live.com
April 11, 11:38 PM
|
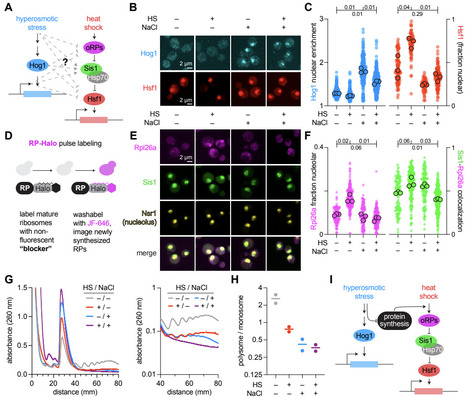
The capacity to adapt to complex environments (adaptability) is a defining property of cells. Yet, its relationship across single-cell physiology, population-level responses, and evolutionary timescales remains unclear. We performed single-cell transcriptional profiling of budding yeast across 20 complex environments before and after long-term selection for increased osmotolerance. In ancestral populations, transcriptional responses organized into a reproducible hierarchy where adaptation to certain cues took precedence over others. This hierarchy mechanistically originated within individual cells through contingent regulation of translation initiation. Evolution for >3,000 generations under osmotic stress increased osmotolerance while reordering this hierarchy, selectively deprioritizing osmotic stress as an organizing axis of adaptation. The derived strain exhibited impaired integration of stress responses, defective translational coordination, and reduced fitness outside the selected condition. Together, these findings illustrate that cellular adaptability is an evolved architecture whose form is set by the history of selection.
|
|
Scooped by
mhryu@live.com
April 11, 11:18 PM
|
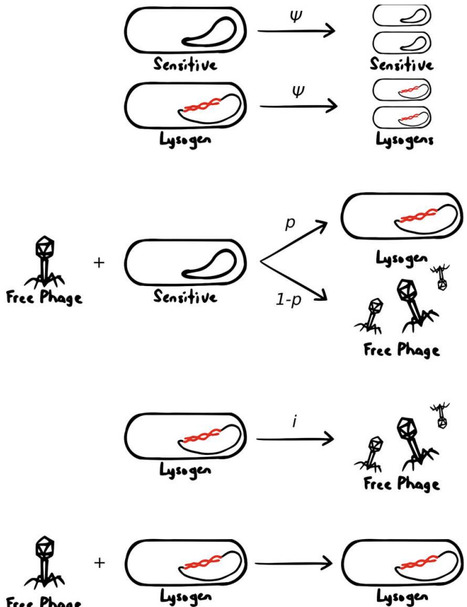
Upon infecting a bacterial cell, temperate phages make a decision between lysis and lysogeny. While research has previously explored how phages sense environmental information to make this choice, most studies have focused on modelling known mechanisms that impact the decision. These mechanisms tell us what environmental information the phage does respond to, but not what it should respond to, as the signals sensed by the phage may serve as proxies for other sources of information. Here, using a mechanism-agnostic population dynamics model, we find that irreversible phage binding to lysogens protects sensitive host cells from infection. This results in lysogens being an additional environmental factor that the phage should sense while making its decision to undergo lysis or lysogeny. Using this model, we derive a responsive lysogeny probability for phages that respond to both cell and lysogen densities optimized towards invading phage-occupied systems, and show that it is more capable of invading and resisting invasion than phage with fixed lysogeny probabilities across different environmental conditions.
|
|
Scooped by
mhryu@live.com
April 11, 5:10 PM
|
Engineering of non-ribosomal peptide synthetase (NRPS) and/or polyketide synthase (PKS) assembly lines to generate modified products has long offered promise to produce novel antibiotics and other bioactive molecules. However, it is only in recent years that this promise has been realised with any consistency. Key to this has been a shift away from engineering approaches informed solely by structural data, and towards strategies that incorporate insights from evolutionary principles and datasets. Such analyses have not only guided the selection of optimal recombination boundaries for substitution of key subdomains, domains or modules, but also methods for increasing engineering throughput, often trading accuracy for volume. Diverse approaches have proven successful in NRPS systems, but a consistent theme has been that recombinant assembly lines are generally impaired in terms of product yield, and a meta-analysis of published results to date indicates that no one engineering strategy is significantly best for minimising yield losses. Evolution-inspired strategies have advanced the engineering of, and product yields for, PKS systems, and further breakthroughs appear imminent. Although no ‘one size fits all’ solution is apparent for either NRPS or PKS engineering, this review highlights important advances in synthetic biology that will support both discovery and production of next-generation antibiotics.
|
|
Scooped by
mhryu@live.com
April 11, 5:07 PM
|
Anaerobic digestion (AD) of food waste (FW) is a key wate-to-energy strategy, yet daily biogas yield is often challenging to sustain, partly due to a limited understanding of the internal methanogens and their functional divergence. Here, we investigated seven full-scale mesophilic FW-AD systems distributed across China along a broad latitudinal gradient (>2,800 km), linking methane production variations (0.38–2.11 m3/m3•d-1) with the phylogenetic distributions of methanogens and their methanogenic genes. We found that hydrogenotrophic and aceticlastic pathways were ubiquitous, whereas methylotrophic methanogenesis showed regional enrichment in warmer regions, reflecting persistent influences of climate-associated upstream conditions on downstream methanogenic communities. Gene-level phylogeny of methanogenesis-related alleles, rather than species-level phylogeny, closely tracked biogas yield variation (Mantel's P < 0.05) and showed consistently stronger associations than gene-level compositions (mean standardized total effect: 0.491 vs. 0.298, P < 0.01). Higher methane yields (1.61 vs. 0.61 m3/m3•d-1 in high- vs. low-performing systems, P < 0.01) were significantly associated with reduced Faith's phylogenetic diversity (1.82 vs. 2.30, P < 0.01) and tighter clustering (mean pairwise phylogenetic distance, MPD: 0.25 vs. 0.30, P < 0.01) of methanogenic gene variants, suggesting that phylogenetic coherence may reflect ecological filtering favoring efficient methanogenesis, albeit at the expense of functional redundancy. These findings highlight gene-level trait phylogeny as a potential proxy for functional robustness, offering a framework for ecological design of AD microbiomes.
|
|
Scooped by
mhryu@live.com
April 11, 4:46 PM
|
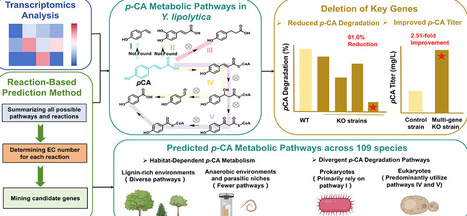
p-Coumaric acid (p-CA) is a universal precursor of plant lignin and polyphenols. A comprehensive understanding of p-CA metabolism is essential for elucidating biological evolution, advancing polyphenol biomanufacturing, and developing lignocellulosic biorefineries. However, p-CA metabolism remains poorly characterized due to the limited number of studies and the absence of universal prediction methods. Here, we systematically elucidate p-CA metabolism in the industrial yeast Yarrowia lipolytica. Through transcriptomic analysis and a reaction-based prediction method, we identify key degradation genes in Y. lipolytica, whose collective deletion reduced p-CA degradation by 81.6% and increased production by 2.51-fold. Comparative analysis of 109 species revealed that the metabolic potential for p-CA is widespread but exhibits significant habitat-dependent divergence. This study establishes a systematic framework for understanding and engineering p-CA metabolism, providing insights for optimizing microbial cell factories and a broader exploration of metabolic pathways.
|














DNA methylation-environment interactions in the human genome