 Your new post is loading...

|
Scooped by
mhryu@live.com
April 9, 11:33 PM
|
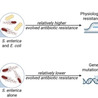
Interspecies interactions can influence the physiology of competing species, shaping their long-term evolutionary trajectories. Although interspecific competition’s role in community dynamics is well-documented, its impact on evolutionary outcomes and mechanisms is less explored. Here, we investigate how interspecies competition affects antibiotic resistance evolution in the gut pathogen Salmonella enterica within synthetic microbial communities. Specifically, we examine how the presence of an interspecific competitor, E. coli, modulates resistance evolution at low streptomycin concentrations. Our findings reveal that interspecies competition results in the selection of S. enterica mutants with higher resistance levels by increasing the likelihood of accumulating resistance mutations that follow a trajectory of negative fitness epistasis. We show that this effect is driven by the enhanced expression of the cryptic aminoglycoside transferase gene (aadA). Our study thus links antibiotic resistance evolution to competition-induced physiological changes, emphasizing the interplay between interspecies interaction and adaptation to environmental conditions.
|
|
Scooped by
mhryu@live.com
April 9, 11:26 PM
|
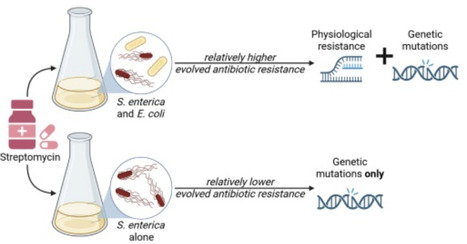
The generation of Metagenome Assembled Genomes (MAG) has become a standard and basic step in the analysis of metagenomic data. This multi-step process, which includes assembly, binning, refinement, and quality control, has many alternative approaches, algorithms, and parameters. Determining the ideal approach for a given ecosystem and study, or highlighting algorithmic gaps in need of additional research and development, requires rigorous benchmarking. We present MAG-E (MAG pipeline Evaluator), a generalizable and expandable framework for end-to-end evaluation of entire MAG pipelines: from assembly, through binning, to quality control and filtering. MAG-E relies on simulations that are built to match an ecosystem of interest and provide a ground truth for accurate evaluation. To demonstrate the capabilities of MAG-E, we benchmark two assemblers, six binning algorithms, three binning modes, and three quality control and refinement methods in the context of the human gut microbiome. Our findings offer multiple insights into optimal MAG generation in this context. We find that metaSPAdes consistently outperforms MEGAHIT in terms of recall (completeness), and that COMEBin overall outperforms alternative binning algorithms, but has lower precision than SemiBin2. While multi-sample binning results in higher precision, as previously shown, single-sample binning has higher recall and leads to better overall performance with modern binners. Binning refinement, which combines bins from multiple different algorithms, leads to reduced performance. We further show that CheckM2 systematically overestimates completeness and underestimates contamination, and that this is partially ameliorated when using GUNC. Finally, we analyze performance at the contig level, and demonstrate that binning algorithms systematically underperform for prophages and fail to bin contigs that are shared between genomes. Overall, MAG-E offers deep insights into successes and gaps in this important analytic process.
|
|
Scooped by
mhryu@live.com
April 9, 11:19 PM
|
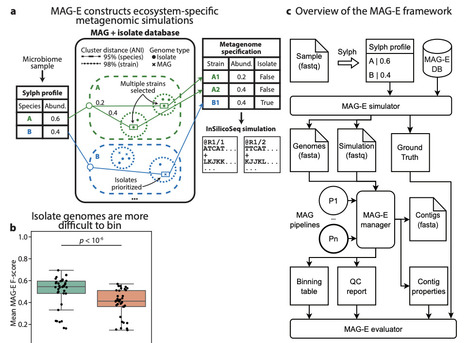
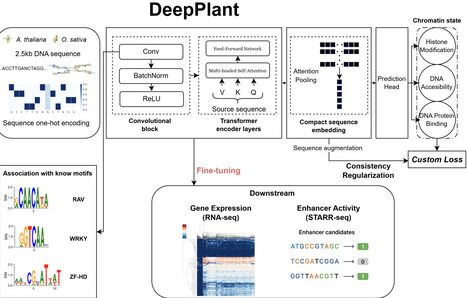
Large-scale sequence-to-function deep learning models have demonstrated unparalleled ability to model biological sequences and have revolutionized the field of regulatory genomics. However, the majority of such efforts have centered on human and mammalian systems, leaving plant regulatory genomics comparatively underexplored. To address this gap, we introduce Deep-Plant, a supervised foundation model trained to predict chromatin state directly from genomic sequence. In contrast to large language models, which are trained in a self-supervised manner using sequence alone, our model is trained to predict chromatin state across tissues and conditions. Training the model on a large collection of genome-wide experiments including DNA accessibility, transcription factor binding, and histone modifications, provides it with added biological context beyond the sequence itself. We demonstrate that the resulting model is an effective platform for developing accurate models of regulatory activity relevant to gene expression and active enhancers, exhibiting large improvements in speed, accuracy, and interpretability over the complementary approach of fine-tuning DNA language models. Deep-Plant models are available in Arabidopsis and rice, and work well as a building block for sequence modeling in related species such as corn. Together, these results establish supervised, chromatin-informed foundation models as a practical and effective paradigm for regulatory sequence modeling in plants.
|
|
Scooped by
mhryu@live.com
April 9, 10:57 PM
|
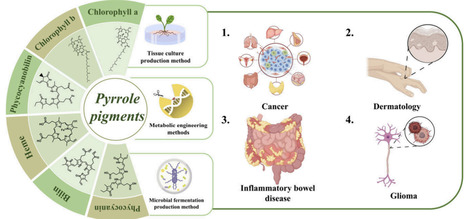
Natural food pigments primarily originate from two sources: chemical synthesis and plant-derived production. With the rapid advancement of society and technology, there is a growing demand for environmentally friendly and healthy food options. Consequently, the demand for safe, nontoxic, and sustainable sources of natural pigments has risen sharply. Natural pigments are biosynthesized during the growth and metabolic processes of plant tissues, and compounds derived from these pigments exhibit a wide range of biological activities that are beneficial to human health and disease treatment. However, due to their inherent instability and low abundance, increasing research efforts have been directed toward the bioengineering of natural pigment production. This review classifies natural pigments into five major structural categories: pyrrole, isoprenoids, quinones, phenols, and betalains. Unlike previous reviews that focused on a single pigment component or specific application fields, this review systematically integrates the biosynthetic pathways, synthetic biology strategies, pharmacological activity mechanisms, and application progress in medicine, health care, and cosmetics of natural pigment-containing medicinal materials. It emphasizes their multiple potentials as “functional pigments” in the development of natural medicines. Additionally, the review combines emerging technologies such as metabolic engineering, artificial intelligence (AI)-assisted screening, and biosensing, proposing a cross-disciplinary development path from basic synthesis to high-value applications and demonstrating strong systematicity and a forward-looking nature. It provides a new integrated perspective for innovative research on natural pigment components.
|
|
Scooped by
mhryu@live.com
April 9, 10:18 PM
|
The Design-Build-Test-Learn (DBTL) cycle forms the basis of modern strain engineering and has accelerated the development of microbial cell factories through increasing automation. Despite significant advances in design and build capabilities, physiology-aware testing and predictive learning remain limited. High-throughput screenings often generate large but shallow datasets that cannot identify mechanistic bottlenecks or ensure robustness under industrial conditions. Furthermore, strain engineering and bioprocess development are frequently treated as sequential rather than integrated activities, leading to scale-up failures and costly late-stage corrections. We propose extending the DBTL framework by treating cell physiology and process constraints as key design variables and integrating automated strain construction, production-relevant phenotyping, and computational models linking genotype, phenotype, and process parameters.
|
|
Scooped by
mhryu@live.com
April 9, 5:39 PM
|
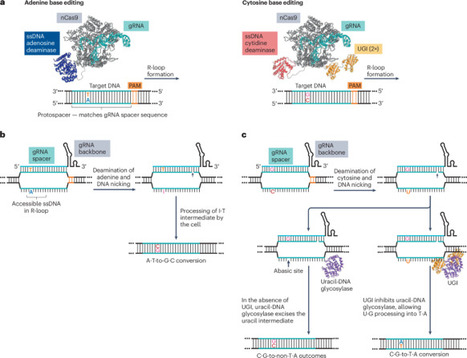
Base editing is a precision genome-editing methodology that enables the programmable installation of point mutations with high efficiency. Two major classes of base editors have been developed: cytosine base editors introducing C·G-to-T·A edits, and adenine base editors introducing A·T-to-G·C edits. Since their introduction, base editor use has expanded substantially, and base editors have been applied in many biological and biomedical applications. In this Primer, we provide an overview of the use of base editors in mammalian cells for high-throughput base-editing screens, disease modelling and therapeutic applications. We cover important considerations in experimental design of base-editing experiments, data analysis and interpretation, and best practices for reporting results. We also discuss potential challenges related to reproducibility and limitations, and outline options for optimization and troubleshooting. Our goal is to aid both new and experienced researchers in effectively implementing base editing in their own work. Base editing enables precise, programmable single-base changes using cytosine or adenine editors. This Primer outlines their use in mammalian cells for screening, disease modelling and therapy, covering experimental design, analysis, limitations and best-practice guidance to support effective implementation.
|
|
Scooped by
mhryu@live.com
April 9, 3:14 PM
|
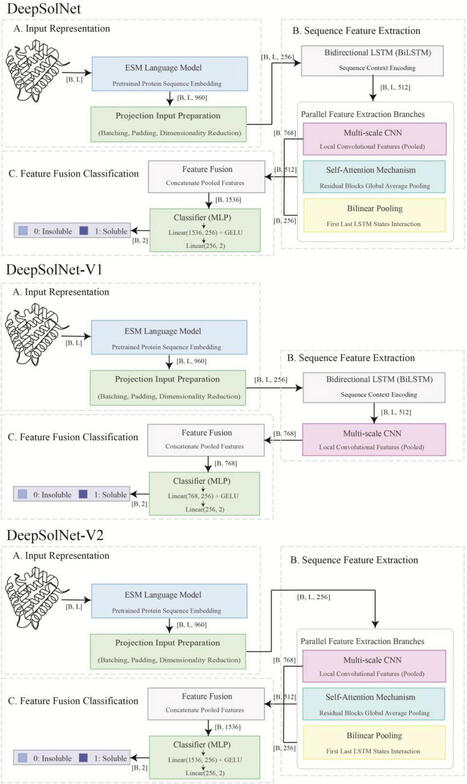
Protein misfolding is a major limitation in prokaryotic expression systems, which lack post-translational modifications and exhibit distinct intracellular environments. This severely hinders the functional expression of many heterologous proteins, especially in E. coli. Accurate prediction of protein solubility is crucial for synthetic biology and protein engineering but remains a challenging task. Here, we present DeepSolNet, a deep learning model that leverages advanced protein language models to enhance solubility prediction. DeepSolNet adopts a multi-module architecture, integrating contextual embeddings from ESM Cambrian with bidirectional long short-term memory networks, convolutional neural networks, and attention mechanisms. On the validation set, DeepSolNet achieved an accuracy of 0.75 and a Matthews correlation coefficient of 0.50 for soluble/insoluble protein classification. On an independently constructed test set containing gammabody, transglutaminase, and aldehyde dehydrogenase sequences, the model maintained high performance with an accuracy of 0.53, achieving state-of-the-art performance. Visualization analyses further showed that DeepSolNet is sensitive to key residues influencing protein solubility. These results demonstrate that DeepSolNet serves as a powerful and generalizable tool for large-scale protein design and expression optimization. The tool is freely available at https://github.com/wangxinglong1990/DeepSolNet.
|
|
Scooped by
mhryu@live.com
April 9, 12:23 AM
|
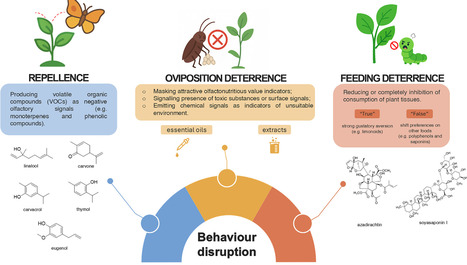
Plant secondary (specialized) metabolites play a pivotal role in disrupting pest behavior, offering a promising and environmentally friendly alternative to conventional pesticides. These compounds can interfere with insect feeding, oviposition, and host selection, thereby reducing crop damage and pest populations. Recent advances highlight the ecological selectivity and rapid biodegradation of these metabolites, making them attractive for sustainable crop protection. Innovative formulation techniques are enhancing their persistence and efficacy, yet challenges remain in understanding synergistic effects, nontarget impacts, and practical implementation. Harnessing the full potential of plant metabolites for pest behavioural disruption requires integrated research and development, paving the way for their broader adoption in integrated pest management strategies.
|
|
Scooped by
mhryu@live.com
April 8, 11:55 PM
|
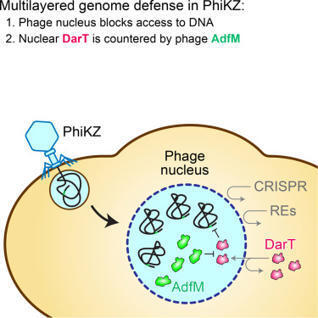
Chimallivirus bacteriophages enclose their replicating genomes in a protein-based compartment termed the phage nucleus. While the phage nucleus segregates phage DNA from host immune proteins, it is not known if additional factors are required to protect against DNA-targeting host defenses. Here, we identify a chimallivirus-encoded DarG2-like antitoxin that localizes to the phage nucleus and provides protection against phage-targeting DarTG2 toxin-antitoxin systems. This protein, which we term AdfM (anti-darT factor macro), contains a macrodomain and removes DarT2-mediated ADP-ribose modifications from DNA. In the absence of AdfM, DarT2 modifies phage DNA and restricts chimallivirus replication despite being largely excluded from the phage nucleus. Increasing the nuclear concentration of DarT2 while decreasing the nuclear concentration of AdfM reduces phage replication. These results show that the phage nucleus is insufficient to completely protect the chimallivirus genome from host defenses; rather, it is one component of a multilayered counter-defense strategy.
|
|
Scooped by
mhryu@live.com
April 8, 11:40 PM
|
Cyclic-di-GMP (c-di-GMP) is a ubiquitous second messenger in bacteria and regulates a variety of cell activities. Many bacteria contain multiple enzymes involved in c-di-GMP synthesis or degradation; however, how they coordinate with each other to orchestrate c-di-GMP homeostasis remains unclear. Here, using the cyanobacterium Anabaena PCC 7120 as a model, we created cdG0 and cdGmax strains by deleting all 8 and 14 genes, respectively, that encode enzymes with c-di-GMP degradation and synthesis domains, alongside a collection of mutants with various numbers of these genes deleted. Our findings demonstrate that c-di-GMP in Anabaena not only modulates cell size but is also indispensable for cell viability. Quantitative analysis established two critical physiological thresholds in vivo: a minimal c-di-GMP level required for cell size maintenance and a lower, lethal threshold essential for survival. We show that the 16 enzymes involved in c-di-GMP turnover in Anabaena function as an electromechanical-like dual relay to control c-di-GMP dynamics, with different modules contributing to c-di-GMP homeostasis or responding as an SOS alarm when the c-di-GMP concentration drops below the lethal threshold. Both effects of c-di-GMP on cell size reduction and cell viability are mediated by the cyclic-di-GMP receptor (CdgR), depending on the amount of the c-di-GMP-free form of CdgR available because of titration by c-di-GMP in the cells. The system, with the two concentration thresholds of c-di-GMP that dictate cell size and viability, respectively, enables dynamic cellular adaptation while preventing lethal effects.
|
|
Scooped by
mhryu@live.com
April 8, 10:52 PM
|
The 3D architecture and dynamics of the genome are crucial for regulation of genome stability, transcription and cellular function. CRISPR-based live imaging technologies have enabled real-time visualization of specific genomic loci and transcripts in living cells. These tools harness customized guide RNAs and nuclease-deactivated Cas effectors to achieve precise genomic targeting, and recent methodological advances provide the 3D spatiotemporal resolution required to decipher real-time chromatin communication. These methods are elucidating the biophysical properties of chromatin, linking dynamic enhancer–promoter interactions directly to transcription, and revealing the role of 3D genome dynamics in basic cellular processes and disease. Here, we summarize the development of CRISPR-based live-cell imaging techniques, highlight the complementary 3D microscopy and analysis methods compatible with these methods, and offer perspectives on their applications to uncover fundamental principles that govern genome dynamics and function. In this Review, Zhu et al. outline the various CRISPR-based tools recently built for dynamic DNA and RNA imaging, which have provided insights into various molecular mechanisms. The authors also discuss the important parameters to consider when using these tools, and how to approach the quantitative image analysis.
|
|
Scooped by
mhryu@live.com
April 8, 10:33 PM
|
Similar to proteins, many RNAs fold into three-dimensional (3D) structures to perform biological functions. Here we present the trRosettaRNA server, a web-based platform for automated RNA 3D structure prediction using deep learning. The primary input is the nucleotide sequence of a target RNA, with the option to upload custom multiple sequence alignments and secondary structures. The server uses an end-to-end neural network for automated 3D structure prediction, followed by an energy optimization step to resolve structural violations. As an automated server, trRosettaRNA is distinguished by its state-of-the-art modeling accuracy, flexible input options and comprehensive visualization of prediction results. trRosettaRNA has been successfully applied in various contexts, including predicting structures for Rfam families lacking known 3D structures, where representative cases of high-confidence structure predictions were found to align well with subsequent experimental observations. Utilizing up to 5 central processing unit (CPU) cores in parallel on our computer cluster, the server takes a median time of about 1 h to predict structures for RNA sequences with about 200 nucleotides. The standalone package for trRosettaRNA offers distinct advantages such as enhanced data privacy for sensitive sequences, the ability to bypass server queues and integration into high-throughput automated pipelines. Importantly, the open-source nature of the package empowers researchers to directly modify the codebase for specialized research needs or to develop derivative tools by fine-tuning the underlying neural network. The web server and standalone package of trRosettaRNA are available at https://yanglab.qd.sdu.edu.cn/trRosettaRNA/ and https://github.com/YangLab-SDU/trRosettaRNA2 , respectively. The trRosettaRNA server is a web-based platform for automated and accurate RNA 3D structure prediction using deep learning. This protocol also describes how to use the standalone package locally, which is beneficial for large-scale applications.
|
|
Scooped by
mhryu@live.com
April 8, 4:54 PM
|
Functional metagenomics has emerged as an effective tool for discovering novel enzymes directly from environmental samples, overcoming the limitations of traditional culture-based methods. In this study, we used a functional metagenomic approach on stool samples from Axis kuhlii, an endemic deer species from Indonesia, to identify active cellulases. We created an efficient workflow for expression of metagenomic sequences directly in Komagatella phaffii by combining metagenomic sequencing to investigate enzyme diversity, multiplex PCR to build a genes library, and rolling circle amplification (RCA) to streamline the cloning process, eliminating the need for intermediate Escherichia coli transformation and propagation steps. Furthermore, a semi-high-throughput screening method was used to evaluate multiple samples at once, allowing for the rapid identification of active enzymes. Using this approach, we discovered five endoglucanases and three β-glucosidases with confirmed enzyme activity. This study shows that functional metagenomics can bridge the gap between computational predictions and experimental validation, providing a reliable platform for enzyme discovery and characterization from complex environmental microbiomes.
|
|
|
Scooped by
mhryu@live.com
April 9, 11:30 PM
|
Scientific breakthroughs infrequently translate into startups without structured training. Stanford’s BioEntrepreneurship Bootcamp teaches a practical framework for navigating bench-to-business translation. The curriculum integrates instruction on intellectual property, team formation, risk assessments, milestone-based funding, and investor communication, with the goal of equipping scientists to advance discoveries into viable ventures.
|
|
Scooped by
mhryu@live.com
April 9, 11:22 PM
|
Generating graphical diagrams of microbial and organellar genomes is a common and essential task in bioinformatics. Existing tools often present a trade-off; while powerful programming libraries that require coding skills, graphical applications require server processing or local installation with complex dependency. This highlights the need for a tool that offers both programmatic control for batch processing and graphical accessibility for ease of use. To fill this gap, I developed gbdraw, a web application that generates circular and linear genome diagrams from self-contained GenBank or DDBJ files or combinations of GFF3 annotation and FASTA sequence files. Its core functions include visualizing annotated features, plotting GC content/skew tracks, and optionally generating pairwise sequence comparisons for comparative genomics. It is available as both a GUI web application and a command-line utility. Unlike existing web-based tools that require data upload to a remote server, gbdraw operates entirely within the user's web browser. This serverless architecture ensures that sensitive sequence data never leaves the local machine, providing a secure environment for visualizing unpublished genomic data. Availability and Implementation: gbdraw is implemented in Python 3 (version 3.10+) and is freely available under the MIT license. The web app is available at https://gbdraw.app/. Source code and documentation are available at https://github.com/satoshikawato/gbdraw. The local version can be installed from the Bioconda channel using a conda-compatible package manager.
|
|
Scooped by
mhryu@live.com
April 9, 11:00 PM
|
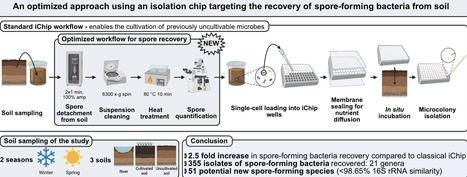
Spore-forming bacteria are key candidates for applications in biotechnology and biocontrol due to their resistance to physical and chemical stress and metabolic diversity. However, their recovery from soil is often hindered by limitations in conventional cultivation methods. The isolation chip (iChip) has expanded access to previously uncultivable microorganisms. This study aimed to optimize pretreatment and incubation conditions for the selective isolation of spore-forming bacteria using a modified iChip protocol. Soil samples were collected from three environments in Brittany, France, during two seasons. Pretreatment included sequential washing, sonication, centrifugation, and heat treatment at 80 °C to select spores while minimising soil particules to allow accurate microscopic quantification. Stainless steel iChip plates with 96 wells were designed to improve membrane sealing and handling. Plates were inoculated with spore-enriched suspensions and incubated in situ for up to six weeks before recovery at the laboratory at two temperatures (15 °C and 30 °C).A total of 757 isolates were obtained, with 52% identified as spore-formers, a substantial increase compared with unmodified iChip workflows (~21%). Taxonomic analysis revealed 79 genera, including Bacillus, Paenibacillus. Phylogenetic analysis highlighted 51 isolates with less than 98.65% 16S rRNA similarity to known species, suggesting potential novelty. The recovery of spore-formers can be achieved through selective pretreatments. Even rare taxa can be revealed with this in situ cultivation. The optimized method offers a promising framework for the recovery of uncultured spore-forming bacteria with potential applications in biocontrol, biotechnology, and natural product discovery.
|
|
Scooped by
mhryu@live.com
April 9, 10:54 PM
|
The ubiquitous presence of Acinetobacter species in the environment holds significant biotechnology potential, particularly in the degradation of various pollutants. In this study, we characterize two plastic-degrading bacteria, strains CAAS 2-6 and CAAS 2-13, isolated from landfill leachate and a strawberry farmland, respectively. Strain CAAS 2-6 showed the highest 16S rRNA sequence similarity (97.7%) with Acinetobacter gerneri DSM 14967T, while strain CAAS 2-13 was most closely related (99.6%) to Acinetobacter kanungonis PS-1T. Phylogenetic analysis of 16S rRNA and gyrB-rpoB genes placed both strains on distinct branches. Genomic comparisons revealed the highest digital DNA-DNA hybridization/average nucleotide identity values for CAAS 2-6 with Acinetobacter indicus CIP 110367T (22.7%/77.0%) and for strain CAAS 2-13 with A. kanungonis PS-1T (66.5%/96.0%). Based on phenotypic and chemotaxonomic data, we propose strain CAAS 2-6T as the novel species Acinetobacter minutum sp. nov. (type strain CAAS 2-6T = GDMCC 1.3951T = JCM 36321T = KCTC 8156T), and strain CAAS 2-13T as the novel subspecies A. kanungonis subsp. fragariae subsp. nov. (strain CAAS 2-13T = GDMCC 1.3956T = JCM 36322T = KCTC 8157T). Both strains utilized polylactic acid, polybutylene succinate-adipic acid (PBSA), polybutylene succinate (PBS), polybutylene adipate terephthalate (PBAT), and polybutylene terephthalate as sole carbon sources, with CAAS 2-6 exhibiting superior growth on PBSA, PBS, and PBAT. Genomic annotation identified genes encoding plastic-degrading enzymes multicopper oxidase AbMCO and alkane hydroxylase AlkB. Enzymatic depolymerization assays confirmed in vitro production of plastic monomers. These findings expand the known metabolic capabilities of Acinetobacter and provide promising new candidates for plastic bioremediation.
|
|
Scooped by
mhryu@live.com
April 9, 9:52 PM
|
Synthetic microbial consortia have been widely used for the production of biochemicals and biofuels. By distributing biosynthetic tasks across multiple strains, it is possible to mitigate metabolic burden, alleviate metabolic crosstalk, and expand substrate and pathway flexibility. To obtain stable microbial consortia, it is crucial to rationally design and control community composition. Here, we review recent advancements in engineering synthetic microbial consortia for biotechnological applications. We also describe the approaches to maintain the stability of synthetic microbial consortia and regulate the populations, highlighting the importance of population control. The future perspective for constructing robust microbial consortia for sustainable biomanufacturing is also discussed.
|
|
Scooped by
mhryu@live.com
April 9, 3:31 PM
|
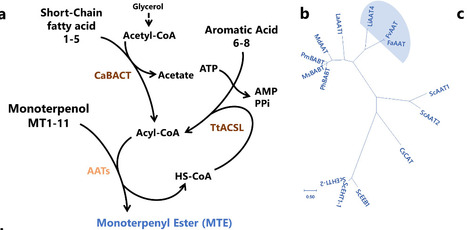
Reconstructing the precise biosynthesis of structurally complex natural esters, such as monoterpene esters, in engineered microbes remains a major challenge, owing to the limited repertoire of highly selective alcohol acyltransferases and the lack of compatible pathway modularity. Here, we establish a dual-substrate microbial platform to profile the activities of alcohol acyltransferase (AAT) to synthesize three distinct classes of monoterpene esters: monoterpenyl esters, monoterpenoate esters, and monoterpenyl monoterpenoate esters, enabling access to both natural and non-natural monoterpene ester biosynthetic pathways. Through structure-guided critical residue engineering and dual-substrate molar ratio tuning, we achieve selective biosynthesis of >C2 acyl-CoA-derived monoterpene esters, despite competing intracellular acetyl-CoA. Coculture engineering further redistributed metabolic fluxes between acyl-CoA and alcohol precursors, yielding 11.50 g/L linalyl acetate and 3.16 g/L geranyl butyrate in 1-L bioreactor. This study expands the biosynthetic space of monoterpene esters and provides a versatile strategy to control AAT selectivity, offering a plug-and-play, scalable framework for ester biomanufacturing. Microbial biosynthesis of monoterpene esters remains a major challenge due to the limited repertoire of highly selective alcohol acyltransferases (AATs). Here the authors discovered and engineered AATs for various monoterpene esters using a dual-substrate microbial platform.
|
|
Scooped by
mhryu@live.com
April 9, 12:35 AM
|
Antagonistic systems of bacteria are often tightly regulated. The human gut Bacteroidales harbor three distinct antagonistic type VI secretion systems (T6SS), one of which is present only in Bacteroides fragilis, known as the GA3 T6SS. Although this is the best studied of the three T6SSs, little is known about how it is regulated. The gene upstream of the GA3 T6SS locus encodes a TetR family transcriptional regulator (TetRGA3), which we show represses expression of the GA3 T6SS locus. The gene immediately upstream and divergently transcribed from tetRGA3, designated here as lgsGA3, encodes a product of the α-oxoamine synthase family of pyridoxal phosphate-dependent enzymes with structural homology to the CqsA autoinducer synthase of the CAI-1 quorum sensing system of Vibrio spp. When lgsGA3 is deleted, transcription of the GA3 T6SS locus is repressed in a TetR-dependent manner. Strains synthesizing LgsGA3 produce a molecule released into the supernatant that likely serves as the TetRGA3 ligand, overcoming TetR transcriptional repression of the GA3 T6SS. We show that GA3 T6SS-specific immunity genes present on two acquired immunity defense islands are also regulated by LgsGA3 coordinating expression of GA3 T6SS antagonism with protection from competitor’s GA3 T6SS toxins. Production and firing of the GA3 T6SS and subsequent antagonism occurs in bacteria deleted for lgsGA3 when growing with bacteria containing this gene or their supernatants or when cocolonizing gnotobiotic mice. These data show that the GA3 T6SS is regulated by a small molecule acting through TetRGA3 allowing the bacteria to coordinate antagonistic and protective systems.
|
|
Scooped by
mhryu@live.com
April 9, 12:12 AM
|
RNA-targeting CRISPR-Cas13 enzymes are robust RNA knockdown tools with both on-target and collateral cleavage activities. However, to date, the in vivo RNA cleavage mechanisms remain poorly understood. Here, we combine in vitro and in vivo methods to elucidate the exact cleavage sites of Cas13. We reveal that some subtypes of Cas13, including Cas13b and Cas13bt, cleave the target RNA at predominant positions, and rational engineering of Cas13 further improves precision. Building on these findings, we develop RNA segment editing (RSE), a targeted RNA cleavage and repair method, to restore dysfunctional RNA in cells. We anticipate that RSE will enable precision RNA engineering for therapeutics and basic research. CRISPR-Cas13 are robust RNA knockdown tools with both on-target and collateral cleavage activities. Here, the authors combine in vitro and in vivo methods to elucidate the exact cleavage sites of Cas13. The authors further develop RNA segment editing to precisely edit dysfunctional RNA in cells.
|
|
Scooped by
mhryu@live.com
April 8, 11:43 PM
|
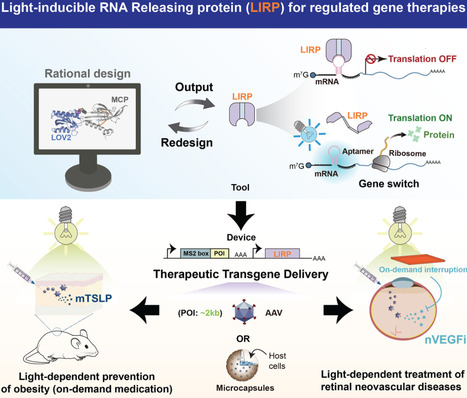
An optogenetic gene switch based on a light-inducible RNA-releasing protein (LIRP) was designed to comply with clinically licensed gene- and cell-based therapy products at a high standard. This LIRP-dependent gene switch is compatible with at least three major administration routes and/or delivery sites in vivo, such as intravitreal and intradermal expression using adeno-associated virus (AAV) vectors, as well as subcutaneous implantation of human induced pluripotent stem cell (iPSC)-derived cell grafts that stably express the corresponding genetic componentry. Using an exemplary case of iPSC-to-hepatocyte conversion, we show that the performance of the light-inducible gene switch was even improved following the differentiation process. This suggests that LIRP could permit robust and consistent regulation profiles across a wide range of cellular environments, which offers the perspective of manufacturing a variety of cell therapy products using different therapeutic chassis. In the particular case of intravitreal expression, a LIRP-dependent gene switch could provide an essential ‘safety upgrade’ for retinal gene therapies, where conventional strategies in the clinics would use AAV vectors to achieve constitutive production of vascular endothelial growth factor (VEGF) inhibitors for the treatment of wet age-related macular degeneration (e.g., NCT05407636). Here, by using the same AAV vectors to deliver a LIRP-dependent gene switch regulating intravitreal expression of similar VEGF inhibitors, we show how a light-regulated treatment regimen could achieve similar therapeutic efficacy as with constitutive expression strategies, but with markedly reduced adverse effects that may result from excessive VEGF inhibition. As LIRP was designed to be compatible with various clinically accepted gene therapy products, it may require minimal safety and efficacy testing as well as modest profitability analyses to establish a new alternative for retinal neovascular disease therapies. Thus, we believe that the overall maturity of the present technology may approach Technology Readiness Level (TRL) 5/6 levels from the viewpoints of intellectual elaboration and overall innovative impact.
|
|
Scooped by
mhryu@live.com
April 8, 11:16 PM
|
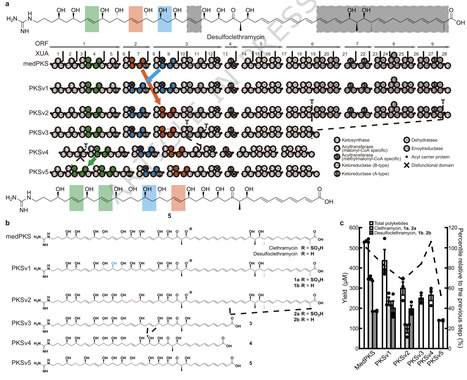
Assembly line biosynthesis creates numerous structurally diverse natural products using a common modular synthetic strategy. The collinearity between the architectures of modular polyketide synthases (PKS) and the structures of their polyketide products would seem to render these biosynthetic machineries excellent platforms for designer biosynthesis, yet reliable strategies to reprogram these assembly lines without diminishing their activities have not been identified. Here, as a best practice for PKS engineering, we demonstrate the reprogramming of the mediomycin PKS without significant loss of productivity. Using in vitro CRISPR/Cas9 gene editing followed by heterologous expression, we reconstruct an inaccessible drug lead of the fibrinogen receptor, tetrafibricin, at 82 ± 3 mg/L yield, retaining 26% productivity after five-step module editing using an evolution-supported cut site, downstream of the acyltransferase domain. A macrocyclic aminopolyol is also accessed through thioesterase swapping. These results pave the way toward the rational reprogramming of PKSs to access desired complex organic molecules. The collinearity between the architectures of modular polyketide synthases (PKS) and the structures of their polyketide products would suggest these biosynthetic machineries are excellent platforms for designer biosynthesis, yet reliable strategies to reprogram these assembly lines without diminishing their activities have not been identified. Here, the authors demonstrate the reprogramming of the mediomycin PKS without significant loss of productivity, and reconstruct an inaccessible drug lead of the fibrinogen receptor, tetrafibricin, at 82 mg/L yield.
|
|
Scooped by
mhryu@live.com
April 8, 10:44 PM
|
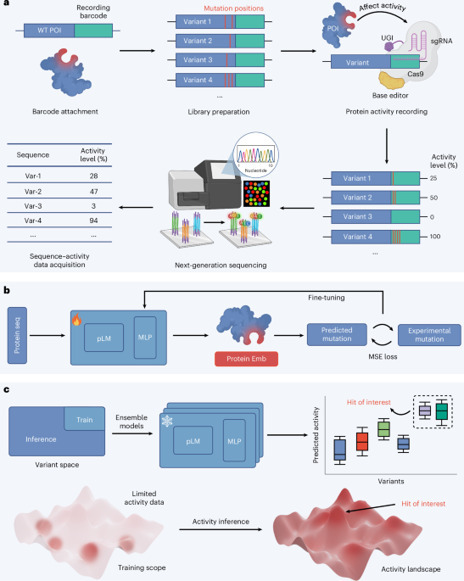
Engineering proteins with desired functions remains challenging and usually requires multiple rounds of screening and selection. Here, we present Sequence Display, a platform that generates large-scale protein sequence–activity datasets in a single round. Sequence Display enables multiplexed assessment of individual variant activity within a single experiment, offering a robust approach to mapping detailed sequence–function relationships. We demonstrate the platform’s broad applicability by generating datasets for cytosine deaminase, uracil glycosylase inhibitor, aminoacyl-tRNA synthetase and a compact Cas9 nuclease. Integrating these datasets obtained from Sequence Display with pretrained protein language models, fine-grained, variant-specific activity landscapes can be constructed. We discovered several Cas9 variants with expanded protospacer-adjacent motif recognition and evolved aminoacyl-tRNA synthetase variants capable of recognizing different noncanonical amino acids. Together, this study establishes Sequence Display as a powerful tool for mapping protein activity landscapes and accelerating the discovery of optimized proteins for biological and medical applications. Sequence Display maps protein variant activities to a sequencing-based readout.
|
|
Scooped by
mhryu@live.com
April 8, 10:30 PM
|
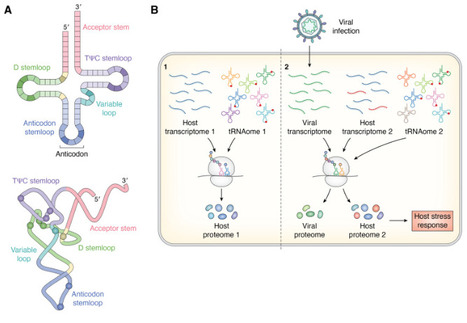
tRNA modifications are key players in post-transcriptional gene expression regulation under external and cellular stresses. Viral infection is a common external stress that hijacks cellular processes for replication. Host cell tRNA pools and modification profiles are often reshaped during viral infection, eliciting both pro- and anti-viral effects. Changes in the host tRNA modifications, particularly in the anticodon sequence, have the capability to reprogram host and viral proteomes. For example, anticodon loop modifications contribute to programmed ribosomal frameshifting essential for producing certain viral proteins. However, the roles of tRNA modifications are not limited to translation during viral infection. Retroviruses use select host cell tRNAs as reverse transcription primers and modifications modulate steps in reverse transcription. Furthermore, some non-primer modified tRNAs are selectively packaged into virion particles, though their functions remain unknown. Virally-encoded tRNAs harbor modifications that expand the anticodon pool, as host tRNAs are depleted. Expression and activity of several tRNA-modifying enzymes are regulated upon viral infection, the functional implications of which remain to be elucidated. tRNA modifications are involved in anti-viral defense, particularly in tRNA cleavage at the anticodon loop following viral infection, leading to tRNA-derived fragments. While the tRNA modification landscape is likely significantly altered following most viral infections, current evidence is limited to a few specific examples. A global tRNAome analysis during viral infection will shed light on the regulation of these and other processes. Emerging technologies including advances in direct tRNA sequencing and modification detection via mass spectrometry are making this possible.
|



















plant gene expression