 Your new post is loading...

|
Scooped by
?
Today, 1:50 AM
|
O-Linked β-N-acetylglucosamine (O-GlcNAc) is an essential nucleocytoplasmic post-translational modification (PTM) installed on many substrates by a single O-GlcNAc transferase (OGT), although functional outcomes for most of these modifications are unknown. Induced proximity methods to write and erase PTMs from desired targets can accelerate functional annotation and identify therapeutic opportunities for PTMs like O-GlcNAc. Here, we report an induced-proximity method with a destabilized nanobody-OGT fusion and demonstrate its general utility for targeted protein O-GlcNAc against 21 substrates followed by annotation of the direct effects of O-GlcNAc on transcription factors in cells. Deeper investigation of AP-1 transcriptional activation reveals an inhibitory nutrient-sensing event regulated by O-GlcNAc on transcription factors c-Fos and c-Jun. Collectively, these data illustrate the rapid investigation of O-GlcNAc functions in cells enabled by a generalizable induced proximity method for targeted protein O-GlcNAc.
|
|
Scooped by
?
Today, 1:28 AM
|
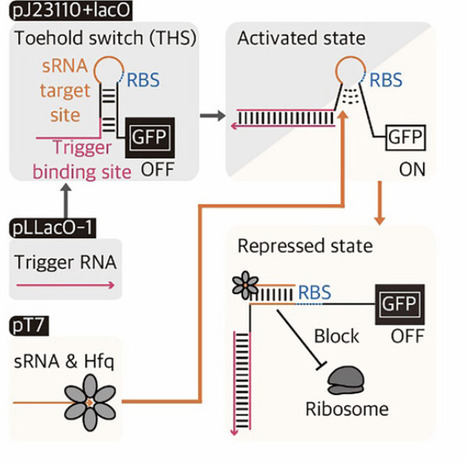
A long-standing goal of synthetic biology is to reprogram cells by rewiring genetic parts. Despite the expanding library of genetic parts, construction of integrated synthetic circuits with desired specifications remains challenging in part due to intricate dependence on sequence contexts, where unexpected narrow dynamic ranges and leaky expression can plague system performance. To provide an alternative approach to the screening process of iterative design-build-test cycles, SUPER (Synthetic Upcycling Platform for Engineering Regulators), a modular platform for upcycling genetic devices is introduced. Inspired by antagonistic regulation mechanisms, SUPER employs small RNA as an add-on controller to modulate gene expression patterns without genetic modification of target regulators. SUPER not only enhances the performance of RNA-, chemical-, temperature-, and protein-responsive regulators up to 1011%, but also allows to cover an expanded dynamic range up to 22 018.9-fold. This enhanced control can provide genetic circuit stability, particularly under strong selective pressures, as demonstrated with a Holin-expressing kill switch integrated with SUPER, maintaining stable functionality for over 30 days. Finally, SUPER combines with an environmental sensor, TlpA36, functioning as a chemical- and temperature-responsive 2-input kill switch. Featuring straightforward design, minimal cellular burden, and expanded tunability, SUPER provides a systematic upcycling framework for genetic circuit construction in biotechnology.
|
|
Scooped by
?
Today, 1:15 AM
|
Viral-mediated bacterial mortality and the prevalence of lysogeny are two key parameters for understanding the role of viral activity in aquatic ecosystems. The viral production assay is most commonly used to assess these parameters, with lytic and mitomycin C-induced viral production rates prevalently extracted using the linear regression or increment-based (VIPCAL) approach. A literature survey shows that 64% of the 89 viral production studies used the linear regression approach for lytic and 48% employed VIPCAL for lysogenic viral production rates. Our comparative evaluation highlights significant differences between these two approaches of estimating viral production rates. To refine estimations, we enhanced VIPCAL to VIPCAL-SE by incorporating standard error of the means to rigorously identify maxima–minima pairs, accounting for biological and ecological variabilities between replicates. We also included a bacterial net generation time endpoint to reduce estimation bias due to potential secondary infections, particularly relevant in more productive ecosystems. VIPCAL-SE is now available as a part of the viralprod R package and provides an opportunity for further standardisation in the field of aquatic viral ecology.
|
|
Scooped by
?
December 15, 11:26 PM
|
With the development of synthetic biology, an evolution system in vivo has been applied to accelerate the construction of cell factories. In this study, an efficient in vivo evolution system was developed for regulation of single and multiple genes in Bacillus amyloliquefaciens. First, the CRISPR/Cas9n-AID base editor was constructed through integration expression of the fused Cas9n protein and activation-induced cytidine deaminase (AID), and the base conversion efficiency from C to T was as high as 90% in single-gene editing. Subsequently, the evolution template (XP43) with an editable RBS sequence (GGGGGGGG) was designed for in vivo evolution through two strategies. By next-generation sequencing of RBS mutation libraries, the extended sgRNA strategy was confirmed to be the optimal evolution scheme. Using the alkaline protease gene (aprE) as the single gene target, the evolution program was initiated to successfully obtain a series of mutant strains with gradient AprE activities. Furthermore, multiple key genes (dhemA, SAM2, and hemEHY) were evolved simultaneously to balance the heme metabolic network, and the optimal mutant strain (HZHA-C2) produced 14.02 mg/L heme, 93% higher than the control strain. Finally, the overexpression of the hemH gene further increased the heme titer by 49%. By a fed-batch fermentation strategy, the heme titer of the optimal engineered strain (HZHA2/pHY-hemH) was improved by 64%, achieving 32.61 mg/L.
|
|
Scooped by
?
December 15, 10:52 PM
|
Chromatin is intrinsically repressive, limiting access to DNA, implying a major regulatory role. Studies with nuclei support this model. However, we have shown previously that genomic DNA is almost completely accessible in living budding yeast and human cells, except for centromeric chromatin. The fission yeast, Schizosaccharomyces pombe, possesses heterochromatin similar to mammalian heterochromatin at the pericentromeric repeats, telomeres and the silenced mating type loci. S. pombe heterochromatin is marked by histone H3K9 di- and tri-methylation (H3K9me2/3) and heterochromatin protein 1 (HP1/Swi6), potentially repressing genes by preventing access to the DNA. Here, we developed a copper-inducible DNA methyltransferase system to measure accessibility in living S. pombe cells. We find that euchromatin and heterochromatin are generally accessible, indicating that heterochromatin does not represent a significant block to DNA methyltransferases in vivo. S. pombe centromeres are much more accessible than budding yeast and human centromeres. In contrast, S. pombe chromatin is mostly inaccessible in isolated nuclei, primarily due to tight nucleosome spacing on gene bodies, with very little linker DNA. We conclude that S. pombe euchromatin and heterochromatin are both highly dynamic in vivo, suggesting that the H3K9me/HP1 system does not repress transcription by preventing access to DNA.
|
|
Scooped by
?
December 15, 10:25 PM
|
Plasmid vectors are to this day the fundamental tools in molecular biology, but their selection is often guided by convenience rather than informed choice. This article revisits the architectural and functional features that determine plasmid performance i.e., origins of replication, copy number, cargo capacity, selection markers, and stability systems. We outline how these elements shape host range, expression dynamics, and metabolic burden, particularly as synthetic biology increasingly targets non-model bacteria. The growing need for reliable, portable vectors has driven the development of broad-host-range backbones, streamlined modular architectures such as SEVA, and alternatives to antibiotic-based selection. We also examine strategies to enhance long-term stability, including toxin–antitoxin systems and chromosomal integration via mini-transposons, recombinase-assisted platforms, and CRISPR-associated transposases. The convergence of standardization and customization, enabled by advances in DNA synthesis and emerging AI-assisted plasmid design tools is discussed also. These innovations promise flexible vector engineering tailored to diverse microbial chassis. Yet, a deeper, systems-level understanding of plasmid–host interactions will be necessary to ensure robust deployment of engineered functions in laboratory, industrial, and environmental settings.
|
|
Scooped by
?
December 15, 10:09 PM
|
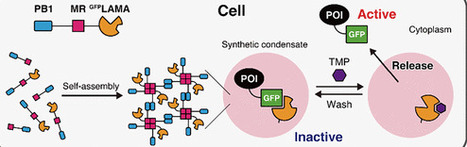
Creating artificial organelles that sequester and release specific proteins in response to a small molecule in mammalian cells is an attractive approach for regulating protein function. In this work, by combining phase-separated condensates formed by the tandem fusion of two oligomeric proteins with a trimethoprim (TMP)-responsive nanobody switch for GFP (GFPLAMA; ligand-modulated antibody fragment), we developed a synthetic condensate system that initially sequesters GFP-tagged proteins within condensates and rapidly releases them into the cytoplasm upon TMP treatment. The released proteins can then be resequestered by washing out the TMP. This system enabled user-defined, temporal, rapid, and reversible control of cellular processes, including membrane ruffling mediated by exogenously expressed GFP-Vav2 and modulation of the cellular localization of endogenous ERK2-GFP generated by genome knock-in. Our results highlight the utility of the GFPLAMA-based synthetic condensate platform as a novel, chemically switchable tool for regulating protein function through controlled protein sequestration and release in mammalian cells.
|
|
Scooped by
?
December 15, 5:44 PM
|
Streptomyces specialized metabolites account for over half of all clinically used antibiotics, as well as numerous antifungal, anticancer, and immunosuppressant agents. Two-component systems, which are widespread in bacteria, are key regulators of antibiotic production in Streptomyces species, yet their activating signals remain poorly understood. CutRS was the first two-component system identified in the genus Streptomyces, and deletion of cutRS in Streptomyces coelicolor was shown to enhance antibiotic production, although its CutR regulon does not include biosynthetic genes. Here, we used Streptomyces venezuelae NRRL B-65442 to further investigate CutRS function. We show that deletion of cutRS increases growth rate and a reversal of the glucose-mediated carbon catabolite repression typically observed in Streptomyces species. We also demonstrate that CutR DNA binding is glucose-dependent, but CutR does not directly regulate genes involved in growth, antibiotic biosynthesis, or glucose metabolism. The only CutR targets conserved in both S. coelicolor and S. venezuelae are the foldase genes htrA3 and htrB, which are involved in the protein secretion stress response. Consistent with this, we show that CutS homologs all contain two conserved cysteine residues in their extracellular sensor domains and that changing these residues to serine constitutively activates S. venezuelae CutRS. We propose that failure of a disulfide bond to form between these cysteine residues indicates secretion stress and leads to activation of the CutRS system and the secretion stress response.
|
|
Scooped by
?
December 15, 5:35 PM
|
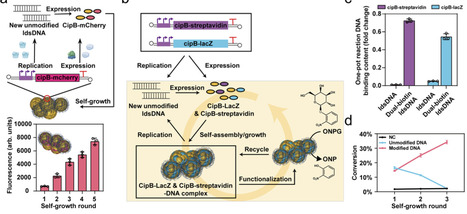
Reconstructing transcription–translation-coupled DNA replication (TTcDR) in artificial systems is crucial for creating synthetic life; however, existing approaches face limitations mainly due to their reliance on purified biological components. Here, we introduce LoopReX, a cell-free system that reconstitutes TTcDR using crude Escherichia coli extracts, offering a more complex native biological environment. LoopReX leverages a minimal machinery composed of phi29 DNA polymerase and T7 RNA polymerase, with the latter facilitating DNA replication initiation through the generation of primer RNAs. Using machine learning, we optimize LoopReX to enhance the efficiency of both DNA replication and protein expression, achieving scalable, sustainable genetic flow and high-yield protein production with robust iterative performance. Furthermore, artificial nucleoids, autonomously formed through CipB-based compartmentalization, improve DNA spatial organization and support multiple biological functions. This work advances the construction of artificial life by reconstituting TTcDR within a single, scalable, and functionalized system, opening exciting possibilities for synthetic biology, biotechnology, and bio-hybrid applications. Reconstructing transcription–translation-coupled DNA replication (TTcDR) in artificial systems is crucial for creating synthetic life. Here the authors develop LoopReX, a TTcDR which leverages minimal machinery and artificial nucleoids to support multiple biological functions.
|
|
Scooped by
?
December 15, 5:22 PM
|
Viral infection of living cells, exemplified by bacteriophage interaction with bacteria, is fundamental to biology and universal across living systems. Here, we establish an all-cell-free viral cycle where T7 phages infect synthetic cells, equipped with lipopolysaccharides on the outer leaflet of the lipid membrane, while encapsulating a cell-free gene expression system. We track each cycle step to demonstrate T7 phage-specific adsorption onto the liposomes, genome entry, replication, expression, and assembly of new infectious virions within the synthetic cells. We quantify key characteristics of the cycle, including the multiplicity of infection, replication efficiency, liposome size constraints, and phage rebinding dynamics. This work establishes a versatile, fully defined in vitro platform for reconstructing and investigating viral infections from individual molecular components. The reconstitution of complex biological processes in cell-free systems can support the detailed characterisation of biochemical mechanisms which are difficult to probe in vivo. Here authors present an all-cell-free T7 phage cycle, consisting of cell-sized liposomes encapsulating a cell-free gene expression reaction and a phage receptor at the membrane.
|
|
Scooped by
?
December 15, 10:59 AM
|
Bacteria and archaea encode on average ten antiphage systems. Quorum sensing, cellular, or transcription factors can regulate specific systems (CRISPR-Cas, CBASS). Yet, a systematic assessment of antiphage systems expression patterns is lacking. Here, we combine publicly available RNA-seq data from 14 different species with an original RNA-seq dataset of 15 Escherichia coli strains across six environmental conditions and two growth stages. Using this data, we explore the transcription patterns of 236 antiphage systems from 81 types. Defense system expression is variable along environmental, physiological, as well as spatial gradients, and can correlate with cellular physiology and mobile genetic element activity. We identify antiphage systems as cohesive but complex transcriptional units, find coordinated expression of defense islands possibly underpinned by local regulators, and demonstrate the functional relevance of differential expression in native systems. Together, these results suggest that environmental and physiological factors regulate prokaryotic immunity and may prime bacteria for infection.
|
|
Scooped by
?
December 15, 10:53 AM
|
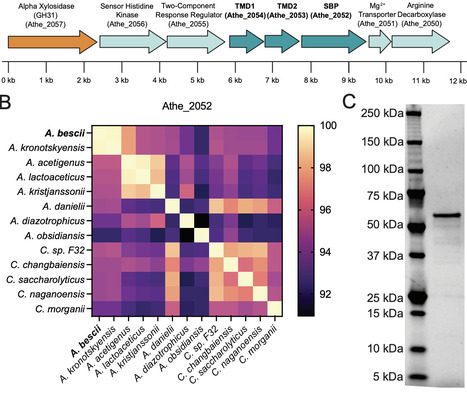
Xyloglucan (an alpha-1,6-xylosyl-substituted beta-1,4-glucan) is a major hemicellulose of the primary cell wall of many plants and an important growth substrate for biomass-degrading bacteria in diverse ecological niches, including the gut microbiome and hot springs. In Gram-positive bacteria, xyloglucan is deconstructed into soluble oligosaccharides in the extracytoplasmic space before import by ATP-Binding Cassette (ABC) transporters, but the structural basis for this process remains poorly understood. Here, we identified an ABC transporter for xyloglucan uptake (Athe_2052-2054) in the Gram-positive, plant biomass-degrading thermophile Anaerocellum bescii, which is conserved across the Anaerocellum genus. We solved the apo crystal structure of its extracellular substrate-binding protein (SBP), Athe_2052, revealing a unique tertiary fold found only in a small subset of SBPs that bind complex oligosaccharides. This structure represents the first ABC SBP known to bind xyloglucan oligosaccharides. Biophysical analysis showed that while Athe_2052 binds unsubstituted beta-glucan chains, recognition of xyloglucan side chains in the binding pocket markedly increases affinity (Kd = 14 nM) for xyloglucan heptasaccharide (XXXG), the principal oligosaccharide released during xyloglucan deconstruction. Molecular modeling revealed that xyloglucan heptasaccharide, owing to its branched substitutions, is bound in a distinct conformation compared to unsubstituted beta-glucans. This represents a unique mode of xyloglucan recognition driven by alpha-linked side-chain interactions rather than beta-glucan backbone recognition alone. Together, these findings provide the first structural basis for xyloglucan oligosaccharide recognition by an ABC transporter in Gram-positive bacteria.
|
|
Scooped by
?
December 15, 10:31 AM
|
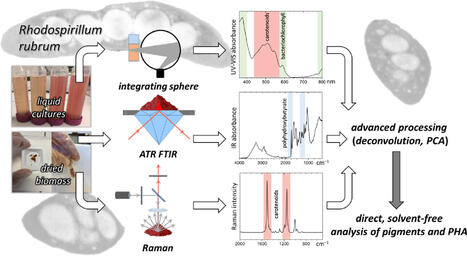
Non-invasive spectroscopic methods are increasingly valued in life sciences, where preserving the native state of biomolecules is essential for accurate interpretation. Traditional analyses of microbial compounds typically involve solvent-based extraction and chromatographic separation processes, which are time consuming, damaging to samples, and can alter biomolecular structures of complexes. To overcome these limitations, we developed a novel spectroscopic workflow for direct metabolite monitoring in microbial cells. Here, we established a combined spectroscopic methodology that allows direct pigment and polyhydroxyalkanoates (PHAs) analysis in complex biological samples without requiring chemical extraction procedures. The UV-Vis spectroscopy technique using an integrating sphere enables direct monitoring of pigments even in turbid whole cell suspensions, providing detailed fingerprints of bacteriochlorophyll a and carotenoids in their natural environment. Together, these techniques provide consistent information about cellular composition. Using the photosynthetic bacterium Rhodospirillum rubrum as a model organism, we demonstrate that our combined spectroscopic approach can resolve pigment states, reveal intracellular PHA content and crystallinity, and measure carotenoids and bacteriochlorophylls directly in native whole cell suspensions. Furthermore, advanced data processing provided an improved interpretation of pigment and PHA states in different cellular forms. This innovative combination of spectroscopic techniques reduces sample manipulation, preserves cellular integrity and provides rapid, precise, and environmentally friendly analysis of microbial metabolites in their natural physiological conditions. The demonstrated workflow is broadly applicable to biological samples where maintaining biomolecular integrity is crucial, and it has strong potential for applications in process analytical technology and industrial biotechnology.
|
|
|
Scooped by
?
Today, 1:37 AM
|
Bacteria are unicellular organisms that typically lack membrane-bound organelles. Nevertheless, they are not merely “bags of enzymes” and instead use alternate mechanisms to organize their components in space and time. Biomolecular condensates are a newly described class of membraneless compartment that organizes cellular functions in bacteria. In this review, we cover key biophysical features of bacterial cells and discuss how their finite size and crowded interior may affect condensate nucleation and stability. Next, we describe three examples of endogenous condensates, highlighting the molecular components driving their formation and the functional roles they may play in cells. Finally, we provide an overview of current and prospective tools to study and manipulate both endogenous and synthetic condensates alike. Overall, bacterial condensates present a fascinating system to explore open questions that span the disciplines of biophysics, molecular and cell biology, and bioengineering.
|
|
Scooped by
?
Today, 1:19 AM
|
Bacterial colonization of tumors is widespread, yet the dynamics during colonization remain underexplored. Here we discover strong variability in the sizes of intratumor bacterial clones and use this variability to infer the mechanisms of colonization. We monitored bacterial population dynamics in murine tumors after introducing millions of genetically barcoded Escherichia coli cells. Results from intravenous injection revealed that roughly a hundred bacteria seeded a tumor and that colonizers underwent rapid, yet highly nonuniform growth. Within a day, bacteria reached a steady-state and then sustained load and clone diversity. Intratumor injections, circumventing colonization bottlenecks, revealed that the nonuniformity persists and that the sizes of bacterial progenies followed a scale-free distribution. Theory suggested that our observations are compatible with a growth model constrained by a local niche load, global resource competition, and noise. Our work provides the first dynamical model of tumor colonization and may allow distinguishing genuine tumor microbiomes from contamination.
|
|
Scooped by
?
December 15, 11:33 PM
|
Small-molecule sensing in plants is dominated by chemical-induced dimerization modules. In the abscisic acid (ABA) system, allosteric receptors recruit phosphatase effectors and achieve nM in vivo responses from µM receptor–ligand interactions. This sensitivity amplification could enable ABA receptors to serve as generic scaffolds for designing small-molecule sensors. To test this, we screened collections of mutant ABA-receptors against 2,726 drugs and other ligands and identified 553 sensors for 6.6% of these ligands. The mutational patterns indicate strong selection for ligand-specific binding pockets. We used these data to develop a sensor design pipeline and isolated sensors for multiple plant natural products, 2,4,6-trinitrotoluene (TNT), and “forever” per- and polyfluoroalkyl substances (PFAS). Thus, the ABA sensor system enables design and isolation of small-molecule sensors with broad chemical scope and antibody-like simplicity.
|
|
Scooped by
?
December 15, 10:57 PM
|
Pseudomonas aeruginosa is a human opportunistic pathogen, capable of producing a wide range of metabolites, including pyomelanin. This pigment results from alterations in tyrosine catabolism. Melanin synthesis from tryptophan has never been reported in Pseudomonas. In this study, we describe a tryptophan-derived melanin in P. aeruginosa PAH, a strain that was isolated from a fibrocystic patient. PAH produced a brown pigment when grown in LB or L-tryptophan-supplemented media. Structural analysis revealed this pigment was composed by two fractions differing in NaOH solubility: a soluble one consistent of pyomelanin, and an insoluble fraction with a complex structure containing substituted indolic units. A pyomelanin inhibitor enhanced total melanin synthesis, mainly the insoluble fraction, and a tryptophan 2,3-dioxygenase inhibitor decreased pigment formation. Metabolomic profiling identified distinct indolic compounds and low levels of anthranilate in PAH cultures. Genomic and transcriptomic analyses revealed the presence of mutations and downregulation of genes related to pyoverdine biosynthesis. Furthermore, iron supplementation in the culture medium reduced melanin production. Overall, tryptophan arises as a key compound for melanin production in PAH, expanding the diversity of melanins synthesized by this genus. Furthermore, iron deprivation emerges as a critical factor triggering melanin biosynthesis, probably as a survival strategy enabling persistence in the fibrocystic lung environment.
|
|
Scooped by
?
December 15, 10:40 PM
|
Several luciferases have been developed for imaging and biosensing, and the collection continues to grow as new applications are pursued. The current workflow for luciferase optimization, while successful, remains laborious and inefficient. Mutant libraries are generated in vitro and screened, “winning” mutants are picked by hand, and the isolated sequences are subjected to additional rounds of mutagenesis and screening. Here, we present a streamlined platform for luciferase engineering that removes the need for manual library generation during each cycle. We purposed an orthogonal DNA replication (OrthoRep) system for continuous hypermutation of a well-known luciferase (GeNL). Short cycles of culturing and screening were sufficient to evolve the enzyme, with no repetitive manual library generation necessary. New GeNL variants were identified that exhibit improved light outputs with a noncognate and inexpensive luciferin. We further characterized the novel luciferases in cell models. Collectively this work establishes OrthoRep and continuous hypermutation as a viable method to engineer luciferases, and sets the stage for more rapid development of bioluminescent reporters.
|
|
Scooped by
?
December 15, 10:24 PM
|
Metabolic regulation─the dynamic biochemical network governing energy transduction, substrate conversion, and metabolic flux─represents a fundamental determinant of microbial viability and functional output. While temporal metabolite fluctuations provide critical insights into metabolic network dynamics, conventional analytical platforms face fundamental limitations in real-time monitoring within native microbial environments. This study presents a novel whole-cell electrochemical biosensing platform integrating carbon dot (CD)-engineered Escherichia coli with advanced cyclic voltammetry (CV) for dynamic metabolic interrogation. This biohybrid system synergizes microbial biochemical specificity with CD-enhanced electron transfer efficiency, achieving nearly a 20-fold amplification in the electrochemical signal amplitude through quantum-enhanced charge transport mechanisms. The platform enables precise quantification of redox-active metabolites via distinct voltammetric fingerprints, as demonstrated through the detection of lactic acid─a pivotal biomarker in industrial biotechnology and clinical diagnostics. Featuring a modular bioarchitectural design, this technology permits seamless adaptation across diverse microbial systems, offering unprecedented capabilities for real-time bioprocess optimization, dynamic metabolic pathway analysis, and pathogen metabolic profiling. By interfacing nanomaterial-enhanced electrochemistry with synthetic biology, our platform surmounts traditional analytical constraints, establishing a versatile analytical tool for spatiotemporal mapping of metabolic networks in complex biological matrices.
|
|
Scooped by
?
December 15, 5:56 PM
|
Despite early assumptions of neutrality, numerous mechanisms are now thought to cause selection on synonymous mutations, commonly supported by a low evolutionary rate at synonymous sites (Ks). This has been best evidenced in the first ~10 codons of genes in E. coli, where Ks is less than around half that of the gene body. Diverse lines of evidence support the hypothesis that these first ~10 codons are under selection for high AT content which causes low mRNA stability that in turn enables ribosomal initiation. There remains one enigmatic discrepancy, however, namely that the low Ks domain extends far beyond the first 10 codons. Here we ask why this is. As we see no evidence that the zone influencing protein levels has been misestimated, we consider three further hypotheses: that reduced Ks is a) owing to overlapping genes, b) reflects an extended slow translational “ramp,” and c) is mutational. We reject the first two as in both Escherichia coli and Bacillus sp. the extended low Ks domain persists on analysis of non-overlapping genes and in Bacillus, where fast optimal codons tend to be A/T-ending, a fast-to-slow codon trend is seen. We fail to falsify the third hypothesis. Employing mutation accumulation data for E. coli we show that the 5′ end has a lower mutation rate, with the first 10 codons having a rate around half that of the gene body, this then steadily increasing following the trend seen for Ks. Compositional variation is likely to explain some of the difference, the 5′ end lacking GC-rich runs while these are most mutagenic. We conclude that even a highly reduced Ks is not always adequate to substantiate selection on synonymous mutations. This result has broad implications for inference of the causes of evolutionary rate variation.
|
|
Scooped by
?
December 15, 5:40 PM
|
Evolution of prokaryotic genomes is highly dynamic, including extensive gene gain via horizontal gene transfer and gene loss, as well as different types of genome rearrangements. Most quantitative analyses of prokaryotic genome evolution are based on single-gene events, although the distribution of genes is known to be non-random at the scales of operons and various genomic islands. Here, we present a spatial-temporal phylogenomic approach for detecting arrays of genes that are likely to have been acquired as a single block. It is shown that the acquisition of multi-gene blocks makes a major contribution to prokaryotic genome evolution and that these blocks consist primarily of co-directed, functionally coherent genes. A detailed analysis of the spatial-temporal data for the genomes of multiple groups of bacteria and archaea shows that the larger blocks of co-acquired genes represent primarily mobile genetic elements (MGEs), in many cases not identified previously. For example, this includes a new group of pleolipoviruses in Haloarchaea and a group of MGEs specific for Bacteroidota with hypervariable gene content and carrying a unique RNA polymerase enzyme. We also show that some ancestral phage-related large islands correspond to previously unnoticed R-type pyocins in Proteus and Morganella genomes. Many of the smaller gene blocks prone to high genome flux are expected to comprise antivirus defense systems and toxins-antitoxins. In a pilot analysis, eight novel toxin-antitoxin and seven novel defense systems were predicted in archaea of the phylum Thermococcaceae.
|
|
Scooped by
?
December 15, 5:26 PM
|
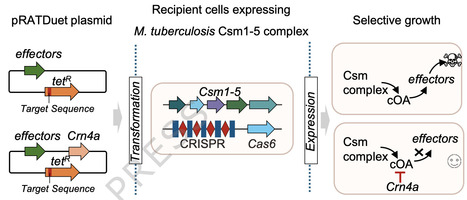
Type III CRISPR systems detect the presence of RNA from mobile genetic elements (MGE) in prokaryotes, providing antiviral immunity. On activation, the catalytic Cas10 subunit conjugates ATP to form cyclic oligoadenylate (cOA) signalling molecules that activate ancillary effectors, providing an immune response. Cellular ring nucleases degrade cOA to reset the system. Here, we describe the structure and mechanism of a new family of ring nucleases, Crn4, associated with type III-D CRISPR systems. The crystal structure of Crn4 reveals a small homodimeric protein with a fold unrelated to any known ring nuclease or, indeed, any known protein structure. Crn4 degrades a wide range of cOA species to linear oligoadenylates in vitro and ameliorates type III CRISPR immunity in vivo. Phage and plasmids also encode Crn4 orthologues that may function as anti-CRISPRs. These observations expand our understanding of ring nucleases and reveal a new protein fold for cyclic nucleotide recognition. Ring nucleases are enzymes that degrade cyclic nucleotide signalling molecules generated by prokaryotic antiviral systems. Chi et al. report the structure and mechanism of a new class of ring nuclease, Crn4, with a very broad substrate specificity.
|
|
Scooped by
?
December 15, 11:56 AM
|
This study establishes a sustainable bioprocess for converting chitosan from marine waste into high-value chitooligosaccharides (COSs), offering an eco-friendly alternative to conventional methods that often generate chemical waste. We achieved heterologous production of chitosanase in an engineered Bacillus subtilis chassis by knocking out its endogenous chitosanase, leveraging the dual advantages of this bacterium as a robust synthetic biology platform and an industrial microorganism. The endogenous chitosanase gene (BsCsn) in Bacillus subtilis WB800N was deleted via CRISPR/Cas9-mediated editing, generating the chassis strain B. subtilis WB800N ΔBsCsn. A codon-optimized GH46 chitosanase (CsnA) from Streptomyces coelicolor, fused to the AprE signal peptide, was then expressed in this host. Response surface methodology optimized the fermentation process, enabling a high extracellular CsnA activity of 540.08 ± 6.20 U/mL, in a 5-L bioreactor under DO-stat-controlled fed-batch conditions. This process achieved a productivity of 11.25 U/(mL·h) and a carbon conversion efficiency of 1682.86 U/g glycerol. Furthermore, MALDI-TOF MS analysis confirmed that CsnA produces COSs with defined degrees of polymerization (DP2-DP4). Conclusion This integrated platform enables the upcycling of marine waste into high-value COSs, establishing B. subtilis as an eco-efficient cell factory and providing a valuable framework for the heterologous expression of other chitosanases in this host.
|
|
Scooped by
?
December 15, 10:56 AM
|
Phylogenetic trees play a fundamental role in elucidating evolutionary relationships among taxa. Clustering taxa remains a major challenge across diverse biological domains such as cancer genomics, microbial systematics, and phylogenomics. Several methods partition taxa in phylogenetic trees into clusters, but these approaches face key limitations. Many rely on arbitrary distance thresholds that are contingent on a-priori information. These thresholds can be hard to ascertain, are subject to bias, and may vary across studies—hindering meaningful interpretation and comparison. More broadly, many methods lack rigorous definitions of what constitutes a cluster, and depend on heuristics that restrict the cluster search space since enumerating all possible clusters is computationally infeasible for large trees. Here, we present PhytClust, a threshold-free algorithm that partitions taxa in phylogenetic trees into monophyletic clusters. PhytClust provides an exact solution to the problem of finding an optimal set of clusters in a phylogenetic tree that minimizes the total intra-cluster branch lengths for a given number of clusters. It then determines the optimal number of clusters using a validity index. PhytClust yields an exact and efficient clustering solution that reflects the tree's topology and genetic distances. In simulated datasets, PhytClust outperforms existing methods in both speed and accuracy and scales to trees with more than a hundred thousand taxa. We apply PhytClust across cancer genomics, avian phylogenomics, bacterial and archaea phylogenetics, and plant genomics to demonstrate PhytClust's varied applicability. By providing a standardized method for taxa clustering within phylogenetic trees, PhytClust yields reproducible, optimal and computationally efficient clusters.
|
|
Scooped by
?
December 15, 10:45 AM
|
Collective behavior is a defining property of multicellular systems, where coordinated outcomes emerge from local cell-cell interactions. Yet the quantitative rules linking single-cell decision-making to tissue-scale organization remain poorly resolved. Here, we develop a quantitative framework that defines an order parameter predicting when initially disordered colonies undergo a transition to ordered fate alignment and when minimal, localized inputs can redirect their collective state. This analysis reveals a distinct control regime in which multicellular assemblies become susceptible to a single engineered guide cell. We validate these predictions by introducing guide cells that integrate into unperturbed colonies and redirect fate patterns within the theoretically defined control windows. Together, these results connect single-cell decision rules to emergent tissue-level organization and establish a generalizable biological control strategy in which a minority engineered subset can reliably redirect the developmental trajectory of a much larger multicellular population.
|



























brockhurst ma,