 Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 1:11 AM
|
In many enzymes, movement of domains from open to closed state forms the environment required for catalysis. We have studied ligand-induced domain motion in 82 enzymes by generating ensembles of AlphaFold 3 (AF3) models both with and without the presence of ligands that are known to trigger such motion. It was found that the results heavily depend on the number of apo and holo structures of each enzyme in the Protein Data Bank (PDB). For enzymes with more apo than holo structures, 64.8% of models generated without ligand are closer to the open apo than to the closed holo state. In contrast, for enzymes that have more holo than apo structures in the PDB, 75.5% of AF3 models without any ligand are in the holo conformation, revealing strong memorization. In both cases, adding the ligand has only a moderate impact. However, the impact of ligand is substantial for proteins that have only a few structures in the training set. Ligands are placed with higher accuracy if there are more holo structures with different ligands in the PDB. We have found that nonbinder ligands also generate similar domain motion, and the distributions of the predicted enzyme conformations remain close to those obtained with the native trigger ligands, but with lower ligand pLDDT values. For enzymes with more holo than apo structures in the PDB, AlphaFold2 also generates the majority of models close to the holo state, suggesting the same memorization effects seen for AF3.
|
|
Scooped by
mhryu@live.com
Today, 12:17 AM
|
Retrons and several defence-associated reverse transcriptases (DRTs) synthesize non-genomic DNA for bacteriophage immunity. In some instances, this non-genomic DNA is of undefined, semirandom sequence. How undefined DNA sequences impart antiphage defence is not known. Here we report the cryo-EM structure and functional characterization of the DRT1 antiphage defence system. We show that DRT1 performs template-free, protein-primed DNA synthesis to generate semirandom DNA adducts. DNA synthesis activates the nitrilase domain of DRT1 while DNA adducts drive assembly of quiescent DRT1 filaments. Filamentous DRT1 is comprised of domain-swapped C-termini that are entwined, forming pseudoknots between tetrameric stacks. This configuration occludes the apo-nitrilase active site, resulting in a dormant state. Bacteriophage escape mutants identify a T4 single-stranded DNA helicase required for DRT1 activity. Functionally, DRT1 resembles a minimal retron where a single gene produces an RT, effector, and non-genomic antitoxin DNA.
|
|
Scooped by
mhryu@live.com
Today, 12:04 AM
|
Dormancy, characterized by an extended lag time before resuming growth, protects bacteria from antibiotic stress and facilitates the emergence of resistance. However, a common mechanism that precisely regulates bacterial lag time remains poorly defined. Here we show that the formation and disassembly dynamics of protein aggregates function as a proteostasis clock that governs the lag time distribution. By tracking the aggregate and regrowth dynamics of thousands of cells, we observed that dormant bacteria from diverse genetic and environmental contexts converge to delayed aggregate disassembly as a common rate-limiting step for growth resumption. Mechanistically, the chromosomal replication initiator DnaA is sequestered in aggregates and spatially separated from the nucleoid, thereby preventing DNA replication until proteostasis restoration enables aggregate disassembly. We modulated bacterial lag time distribution by genetically and chemically targeting the proteostasis-replication axis. Remarkably, lag extension scales linearly with the disruption level of proteostasis across diverse backgrounds, indicating a universal coupling between dormancy and proteostasis. This proteostasis collapse renders dormant bacteria, including clinical isolates, more sensitive to additional proteotoxic stress. These findings reveal a timekeeping mechanism in cellular dormancy, highlighting proteostasis as a shared target for combating antibiotic tolerance.
|
|
Scooped by
mhryu@live.com
March 9, 10:03 PM
|
Ribo-Seq libraries often contain highly abundant non-coding RNA contaminants, which are challenging to remove due to their high sequence variability and diverse fragmentation patterns. We present an organism-independent computational pipeline that identifies experiment-specific target sequences and enables their efficient depletion using custom-tailored LNA probes in a single pipetting step. We demonstrate that LNA-based depletion is most effective during library amplification and has no effect on gene-level quantification. Contaminant depletion in Arabidopsis libraries nearly doubled the yield of coding reads, significantly improving cost-effectiveness.
|
|
Scooped by
mhryu@live.com
March 9, 3:40 PM
|
Bacteria and fungi ubiquitously coexist, with their interactions critically influencing human health and industrial processes. Quorum sensing (QS) is a core regulatory mechanism that enables density-dependent coordination and phenotypic responses across these two kingdoms. While bacteria and fungi utilize their respective QS systems to engage in competitive or cooperative interactions to enhance their environmental adaptability, the current understanding of QS-based communications between them remains scattered, and a systematic summary of this field is still lacking. In this review, we examine the intricate dialog between bacteria and fungi, focusing on its role in microbial network assembly and ecosystem function, to provide a comprehensive analysis and engineering perspective on QS-based cross-kingdom communication. Specifically, we will first briefly delineate the core architecture of bacterial and fungal QS systems and the phenotypes they govern. Then, we will analyze QS-based interactions across diverse environments between different bacteria and fungi, categorizing natural QS interactions based on various phenotypes, including biofilm co-assembly and metabolic complementation. We further compare and analyze synthetic biology strategies, including promoter engineering and directed evolution of QS regulatory components, for reprogramming bacterial-fungal interactions and their applications. By synthesizing and contrasting these natural paradigms with synthetic designs, we provide a blueprint for achieving modular control over bacterial-fungal communities in diverse environments. Finally, by outlining persistent challenges and future trends, we aim to propel this field forward, enabling the deciphering of complex microbial interactions and ultimately increasing our capacity to engineer microbial consortia for diverse applications.
|
|
Scooped by
mhryu@live.com
March 9, 3:24 PM
|
Microbial competition for trace metals shapes their communities and interactions with humans and plants. Many bacteria scavenge trace metals with metallophores, small molecules that chelate environmental metal ions. Metallophore production may be predicted by genome mining, where genomes are scanned for homologs of known biosynthetic gene clusters (BGC). However, accurately detecting non-ribosomal peptide (NRP) metallophore biosynthesis requires expert manual inspection, stymieing large-scale investigations. Here, we introduce automated identification of NRP metallophore BGCs through a comprehensive algorithm, implemented in antiSMASH, that detects chelator biosynthesis genes with 97% precision and 78% recall against manual curation. We showcase the utility of the detection algorithm by experimentally characterizing metallophores from several taxa. High-throughput NRP metallophore BGC detection enabled metallophore detection across 69,929 genomes spanning the bacterial kingdom. We predict that 25% of all bacterial non-ribosomal peptide synthetases NRPS encode metallophore production and that significant chemical diversity remains undiscovered. A reconstructed evolutionary history of NRP metallophores supports that some chelating groups may predate the Great Oxygenation Event. The inclusion of NRP metallophore detection in antiSMASH will aid non-expert researchers and continue to facilitate large-scale investigations into metallophore biology.
|
|
Scooped by
mhryu@live.com
March 9, 2:56 PM
|
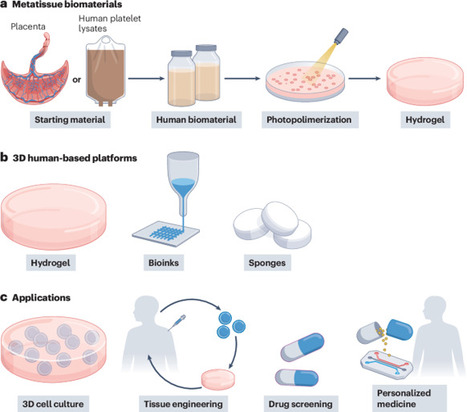
Human protein-based biomaterials facilitate the development of preclinical models that integrate predictive value and ethical responsibility. Metatissue uses decellularized extracellular matrix from placenta samples and human blood to create tunable, xeno-free substrates for 3D cell culture, minimizing the reliance on animal models and accelerating translational research.
|
|
Scooped by
mhryu@live.com
March 9, 2:43 PM
|
Nucleic acid degradation is a common strategy for prokaryotic antiphage systems, as exemplified by the CRISPR-Cas system. The PD-(D/E)-XK nucleases constitute a widely distributed family in these defenses. Notably, most members exhibit a single nuclease domain, while variants containing dual nuclease domains within a single polypeptide remain underexplored, and their molecular mechanisms largely obscure. Here, we biochemically and functionally study a single-protein system containing an uncharacterized PD-(D/E)-XK defense protein (Upx). As revealed by single-particle electron cryo-microscopy (cryo-EM) structure, the C-terminal domain (CTD) harboring the conserved PD-(D/E)XK catalytic core is buttressed by the N-terminal domain (NTD) and the middle domain (MD). Functional assays demonstrate that the nucleic acid binding capability of the CTD is enhanced by the MD. The NTD also displays a noncanonical, basal exonuclease activity that is auto-inhibited by MD. IP-MS experiments identify Upx-interacting phage proteins, and substrate profiling defines its physiological preferences, collectively pointing to its potential physiological targets. Notably, the phage protein gp16 was found to relieve MD-mediated inhibition of the NTD, suggesting a virus-triggered mechanism for activating Upx’s dual nuclease activity. Together, these findings establish Upx as a single-protein dual-nuclease anti-phage system, expanding our understanding of bacterial immunity and informing antiviral strategy development. Bacteria employ diverse nucleases to defend against viruses. Here, authors characterize Upx, a single-protein, dual-nuclease system, and reveal that a specific phage protein relieves auto-inhibition to activate its antiviral function.
|
|
Scooped by
mhryu@live.com
March 9, 12:58 PM
|
Adipic acid (AA) is an important dicarboxylic acid that serves as a precursor in the synthesis of nylon-6,6. Given the increasing market demand for AA and the environmental concerns associated with its conventional production, the development of sustainable biosynthetic techniques for AA production has become a key research focus in both industry and academia. However, industrially viable technologies for AA biosynthesis remain constrained by several challenges, particularly incomplete raw material utilization, low strain conversion efficiency, a complex fermentation process, and high costs of downstream separation. To overcome these barriers, this review presents the current state of AA biosynthesis, critically discussing biosynthetic pathways and advanced metabolic engineering strategies and tools for constructing cell factories with high conversion efficiency. The basic principles relevant to improving the fermentation process and downstream separation technologies are also comprehensively reviewed. Key challenges and knowledge gaps are identified, providing insights to guide future research toward commercially viable biobased AA production.
|
|
Scooped by
mhryu@live.com
March 9, 11:01 AM
|
Biological research often involves complex, repetitive, and high-throughput manipulations that are well-suited to automation. However, current robotic systems generally excel only at narrowly defined tasks or standardized workflows and remain expensive, inflexible, and dependent on proprietary modules or reagents. To address these limitations, we developed the Open-Source Collaborative Automation & Robotics (OSCAR) platform, a flexible and low-cost system designed to perform common laboratory manipulations using standard, human-operated equipment. OSCAR incorporates open-source software and modular hardware to maximize accessibility and affordability. The platform features a robotic arm equipped with a dual-function end-effector: a pipetting module for precise liquid handling and a vision-enabled gripper for manipulating laboratory tools. To demonstrate the platform’s versatility, we implemented a representative plasmid assembly workflow, from PCR amplification and enzymatic assembly to transformation, plating, colony picking, PCR screening, and validation by agarose gel electrophoresis. By making this system open-source and compatible with widely used consumables and equipment, we aim to democratize access to automation and broaden its adoption across diverse research environments.
|
|
Scooped by
mhryu@live.com
March 9, 10:36 AM
|
Insulin glargine, a long-acting insulin analog, is essential for diabetes treatment. However, its industrial production remains challenging due to limitations in conventional expression systems. Here, we employed the industrial filamentous fungus Trichoderma reesei as an alternative expression host for insulin glargine production, leveraging its superior protein secretion capacity. Initially, expression constructs containing the constitutive Pcdna1 promoter and CBH1 signal peptide (SP1) showed successful transcription but failed to achieve extracellular secretion, presumably due to induced endoplasmic reticulum (ER) stress. Consequently, we implemented a fusion protein strategy utilizing three distinct carrier proteins (CBH1, CBH2, and LA-20) to enhance glargine secretion. Notably, the CBH1 fusion not only enabled detectable glargine secretion but also significantly alleviated the ER stress. Furthermore, replacement of SP1 with the Aspergillus niger β-glucoamylase (glaA) signal peptide achieved a fourfold enhancement in glargine secretion and further reduced cellular stress responses. Following these systematic optimizations, a final yield of 58.95 mIU/L glargine was achieved in shake-flask cultures. Thus, the combined strategy described here could achieve extracellular production of glargine in T. reesei, suggesting that it is a promising host for secretory production of therapeutic recombinant proteins, particularly complex analogs like glargine.
|
|
Scooped by
mhryu@live.com
March 9, 10:12 AM
|
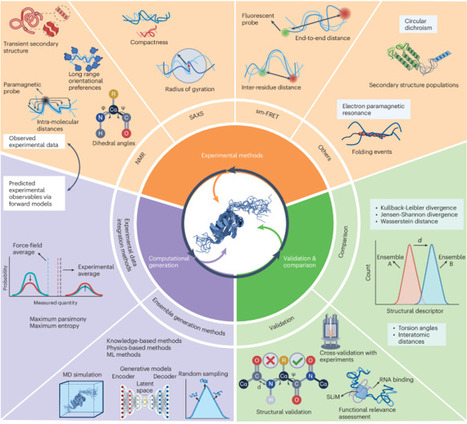
Disordered proteins play essential roles in myriad cellular processes, yet their structural characterization remains a major challenge due to their dynamic and heterogeneous nature. Here we present a community-driven initiative to address this problem by advocating a unified framework for determining conformational ensembles of disordered proteins. Our aim is to integrate state-of-the-art experimental techniques with advanced computational methods, including knowledge-based sampling, enhanced molecular dynamics and machine learning models. The modular framework comprises three interconnected components: experimental data acquisition, computational ensemble generation and validation. The systematic development of this framework will ensure the accurate and reproducible determination of conformational ensembles of disordered proteins. We highlight the open challenges necessary to achieve this goal, including force-field accuracy, efficient sampling, and environmental dependence, advocating for collaborative benchmarking and standardized protocols. This Perspective establishes a comprehensive and practical framework to guide intrinsically disordered protein (IDP) ensemble determination, benchmarking and interpretation, as well as proposes a roadmap for IDP ensemble determination, uncertainty quantification and actionable benchmarking strategies.
|
|
Scooped by
mhryu@live.com
March 9, 9:30 AM
|
Aspergillus fumigatus is the leading cause of invasive mold infections in immunocompromised patients. Current antifungal treatment primarily depends on the triazole antifungals, which act by inhibiting Erg11/Cyp51, a key enzyme in the ergosterol biosynthetic pathway. However, resistance is emerging at an increasing rate, reducing treatment efficacy and patient survival. Confirmed resistance mechanisms in clinical isolates include mutations in cyp51A, cyp51B, hmg1, hapE, cox10, and the overexpression of drug efflux pumps. To identify additional determinants of triazole resistance, we grew A. fumigatus wild-type and Δcyp51A mutant strains under increasing concentrations of voriconazole. Sequencing of the resultant resistant strains identified known mutations in cyp51A and cyp51B, and novel mutations in hmg1, abcC (cdr1B), ptaB, erg25B, and srbA. Mutations of hmg1 and ptaB occurred early during evolution, while mutations of erg25B and srbA occurred later. Reintroduction of the novel mutations in hmg1, abcC, ptaB, and erg25B into wild-type A. fumigatus and correction of the srbA mutation in the evolved strain validated their contribution toward triazole resistance. Sterol profiling analysis indicated that mutation or deletion of erg25B is associated with a decrease in the accumulation of methylated sterols. Mutation or deletion of ptaB resulted in increased cyp51A, cyp51B, and erg25A expression. Sequence analysis of clinical isolates revealed enrichment of missense mutations in ptaB, hmg1, abcC, and srbA among triazole-resistant strains.
|
|
|
Scooped by
mhryu@live.com
Today, 1:07 AM
|
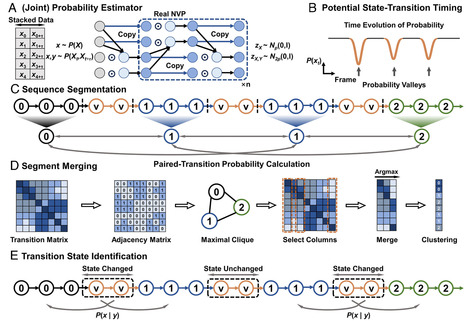
Revealing the complex mechanisms of protein folding, including the transient intermediate states that govern the process, is a fundamental goal in computational biophysics. While molecular dynamics (MD) simulations generate vast amounts of data to this end, extracting a clear kinetic model from these complex, high-dimensional trajectories remains a significant challenge. We present AI-Based conditional transition clustering (CTC), a framework for analyzing MD trajectories that directly addresses the limitations of state-centric methods. Conventional approaches, such as Markov state models, rely on predefined geometric clustering or assume fixed linear dynamics, which can bias the discovery of protein conformational states. CTC operates on a “dynamics-centric” principle, defining a conformational state as a kinetically trapped region identified after analyzing the system dynamics, not before. By leveraging AI-based normalizing flows to estimate conditional transition probabilities from the MD data, CTC identifies states as “kinetic islands” with low escape probabilities. Applying CTC to protein-folding simulations successfully identifies critical intermediate and transition states, revealing folding pathways without prior assumptions about the number of states or their kinetic properties. This approach provides a more objective and physically grounded method for uncovering the complex mechanisms of biomolecular systems.
|
|
Scooped by
mhryu@live.com
Today, 12:06 AM
|
Antimicrobial-resistant Staphylococcus aureus and S. epidermidis remain major causes of invasive infection, yet isolation of therapeutically robust lytic phages targeting Gram-positive pathogens is often constrained by adsorption barriers imposed by thick peptidoglycan cell walls. We demonstrate that these limitations are primarily methodological. By optimizing the ionic microenvironment during isolation through divalent cation supplementation (10 mM CaCl2 and MgSO4), controlled enrichment, phage polyclonal mixtures, and limited lysozyme-assisted release, we recovered 28 genomically distinct lytic phages compared with six obtained using conventional protocols. Host-range profiling across 103 clinical isolates showed broad infectivity, with 51.7% of interactions supporting high-efficiency infection and multiple phages exhibiting cross-species activity. Ion-supplemented adaptive passaging restored and expanded lytic capacity against resistant strains within five rounds. These findings show that ionic regulation of adsorption reshapes Gram-positive phage-host dynamics and provide a scalable framework for precision targeting of resistant Staphylococcus infections.
|
|
Scooped by
mhryu@live.com
March 9, 10:45 PM
|
The bacterial actin-homolog MreB is a crucial component of the Rod-system (elongasome) that maintains rod shape in many bacteria. It is localized beneath the cytoplasmic membrane, where it organizes the elongasome complex. Depletion or deletion of mreB results in loss of rod shape and cell death; however, the mechanism of how MreB operates is not known. Past studies have reported that mutations in mreB cause varying degrees of cell shape and size alterations based on the type and position of the substitution. To better understand the role of MreB in rod shape formation we have taken the first truly systematic approach by replacing the native copy of mreB with an alanine-scanning mutagenesis library. Surprisingly, we observed stably growing spherical mutants that have lost MreB’s function(s) for shape regulation without losing viability. Hence, MreB has vital functions related to growth in addition to shape maintenance that can be separated. In support of this, rod shape suppressor analysis of these spherical mutants only revealed reversions or intragenic mreB mutations, suggesting that MreB is indispensable for rod shape. Additionally, our results imply the elongasome is no longer active in these strains, suggesting a novel way for rod shaped bacteria to synthesize cell wall.
|
|
Scooped by
mhryu@live.com
March 9, 3:56 PM
|
Genome engineering plays a crucial role in the rapidly growing fields of metabolic engineering and synthetic biology. Chromosomal integration and stable expression of functional genes or large metabolic pathways necessitate the development of host-independent enabling technologies in diverse bacteria. Here, a generalizable genome engineering approach, MNGE (Multi-targeting Non-specific Genome Engineering), is developed based on the multi-targeting integrase (MTI) systems for multi-copy (at least three copies), highly random (only requiring the core TT dinucleotide) integration of metabolic genes or pathways in both Gram-positive bacteria (Streptomyces and Saccharopolyspora) and Gram-negative bacteria (Burkholderia and Chromobacterium). Using MNGE, the fungicide UK-2 BGC (41 kb) and the polyether antibiotic salinomycin BGC (106 kb) were randomly integrated into a heterologous host Streptomyces albus, significantly enhancing their fermentation levels based on chromosome position effects. Furthermore, the potent Gq/11-signaling inhibitor FR900359 BGC (66 kb) was successfully expressed in Burkholderia gladioli by the MTI1 system. Together, the MNGE approach exhibits broad applicability for next-generation genome engineering in diverse bacteria, thereby achieving highly efficient production of high-value compounds.
|
|
Scooped by
mhryu@live.com
March 9, 3:35 PM
|
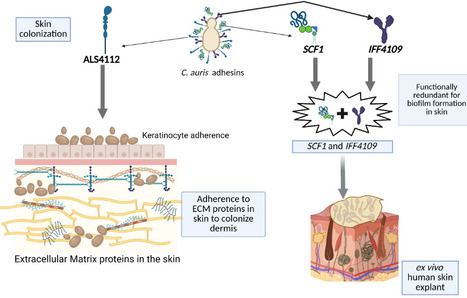
Candidozyma auris (formerly Candida auris) is an emerging multidrug-resistant fungal pathogen that causes life-threatening infections in humans. C. auris is distinct from other Candida species and exhibits exceptional capacity for skin colonization, resulting in nosocomial transmission and outbreaks of invasive infections. Fungal adhesins play a crucial role in skin colonization. With this perspective, we discuss the recent advances in the fungal adhesins of C. auris and how the divergence of adhesins in C. auris contributes to its unique fitness for skin colonization. We also discuss potential avenues to target fungal adhesins, which could pave the way for developing novel vaccine strategies and therapeutics to prevent skin colonization, nosocomial transmission, and invasive C. auris infections in humans.
|
|
Scooped by
mhryu@live.com
March 9, 3:02 PM
|
Scientists say they have made some of the first direct measurements of how long it takes an individual, ordinary protein to fold. The results were surprising: they found no relationship between a protein’s sequence or size and how long it takes to fold into its 3D shape. And proteins seem to fold more efficiently than do other biomolecules, such as DNA — despite proteins having a more complex set of ingredients. The work was published today in Physical Review Letters. The authors attached a red dye molecule to one end of a string of amino acids, and a green one to the other end. The green dye shines on its own. The red dye is activated only when it receives energy from the green dye. Before the amino-acid string folds, the fluorescence from the green dye is visible. When the string starts folding, the two dye molecules are brought closer together, allowing energy to transfer from the green molecule to the red molecule, which then begins to shine. The fastest transition-path time was less than a microsecond, and the slowest was about four microseconds.
|
|
Scooped by
mhryu@live.com
March 9, 2:54 PM
|
Developing short, stable, and potent antimicrobial peptides is a promising strategy to combat antibiotic resistance and persistence. We present CAMPER (Constraint-driven AMP Engineering with Ranking), a mechanistic artificial intelligence framework that integrates machine learning with biophysical ranking to prioritize membrane-targeting peptides effective against persister and biofilm forms of methicillin-resistant Staphylococcus aureus. We apply CAMPER to identify WP-CAMPER1 (12mer) that kills S. aureus MW2 at a minimal inhibitory concentration of 4 µg/mL. A 2% topical WP-CAMPER1 formulation reduces S. aureus MW2 burden by 2.5 log10 (p < 0.0002) in a murine prophylactic skin infection model, while its D-enantiomer, WP-CAMPER1-d, achieves 1.37 log10 (p < 0.0001) reduction in an established biofilm infection model. Single-cell analysis using a high-throughput microfluidic system shows that WP-CAMPER1-d reduces exponential-phase persisters of S. aureus USA300, and, in a deep-seated murine thigh infection model, decreases stationary-phase S. aureus MW2 persisters by 1.6 log10 (p < 0.0001). This study introduces CAMPER, a mechanistic artificial intelligence platform for designing antimicrobial peptides targeting MRSA. CAMPER identified a stable peptide that eradicates MRSA biofilms and persister cells and was active in mouse infection models.
|
|
Scooped by
mhryu@live.com
March 9, 2:29 PM
|
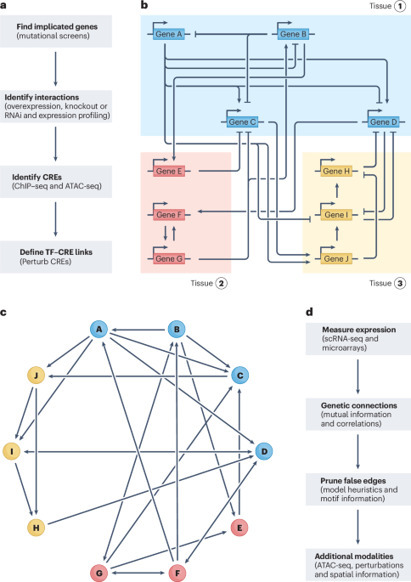
Gene regulatory networks (GRNs) explain how the genome controls cellular behavior and tissue morphogenesis, serving to connect molecular mechanism to functional output. Single-cell technologies now provide descriptions of these networks with unprecedented detail, but this advance has also revealed gene regulatory systems that are too complex for our existing conceptual frameworks. GRNs, which should provide mechanistic explanations, are increasingly reduced to statistical correlations — ‘hairballs’ that fail to capture molecular causation. Here, we explore why this dilemma exists and propose a path forward. We argue that methods in ‘representation learning’ can be used to model GRNs, without needing to capture every molecular detail. For this framework, we advocate three linked principles: models must be inherently mechanistic, with structures grounded in cellular and evolutionary biology; molecular principles and constraints must be used to reduce the solution space for learning GRN models; and more sophisticated forms of experimental perturbation and synthetic biological engineering are needed to train models and test predictions. By reimagining GRNs through these principles, we can bridge the gap from data abundance to new conceptual understanding. In this Perspective, Maizels and Briscoe discuss the limitations of current models of gene regulatory networks and outline solutions to harness data abundance without compromising explanatory power.
|
|
Scooped by
mhryu@live.com
March 9, 12:54 PM
|
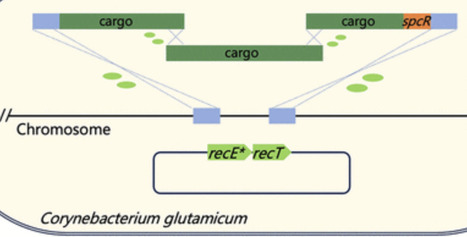
Corynebacterium glutamicum is a key industrial chassis for producing high-value chemicals, particularly amino acids. However, integration of large DNA fragments remains either inefficient or labor-intensive. Here, we optimized a RecET variant-assisted homologous recombination system to achieve single-step integration of DNA fragments larger than 11.3 kb by transforming donor DNA derived from either chromosome or linear DNA fragments. To further expand the size limit, we developed the One-Step Multi-Fragment Assembly Integration (OMAI) strategy, in which multiple overlapping PCR fragments are cotransformed and assembled in vivo, permitting integration of heterologous sequences with a length approximately 50% longer than the conventional single-fragment limit. Each editing cycle of OMAI is completed within 3 days, which is expected to be the most rapid method for large-fragment insertion in C. glutamicum.
|
|
Scooped by
mhryu@live.com
March 9, 10:55 AM
|
Droplet digital (dd) CRISPR integrates the high sequence specificity of CRISPR-based nucleic acid detection with the absolute quantification capability of digital droplet microfluidics, offering high sensitivity, precision, and scalability. By partitioning samples into thousands to millions of picoliter microdroplets, ddCRISPR enables single-molecule resolution and minimizes background interference. This review summarizes the principles of droplet generation, manipulation, and detection in ddCRISPR platforms, as well as recent advances in amplification-based and amplification-free detection strategies. Representative applications are highlighted for viral, bacterial, and other DNA/RNA biomarker detection. Current challenges, including workflow automation, droplet stability, multiplexing, and assay portability, are discussed alongside future perspectives such as artificial intelligence (AI)-assisted analysis, point-of-care integration, and high-throughput multiplexed detection. These insights aim to guide the translation of ddCRISPR technologies from laboratory research to robust, scalable, and accessible diagnostic solutions.
|
|
Scooped by
mhryu@live.com
March 9, 10:16 AM
|
Within lignocellulosic biomass, xylose is the second most abundant sugar after glucose. As a renewable and sustainable substrate, it is gaining attention as a feedstock for microbial bioprocesses. In this study, we demonstrated the co-production of polyhydroxybutyrate (PHB) and violacein from xylose. We initially confirmed the feasibility of co-production through genome-scale metabolic simulations, followed by optimization using a hybrid expression system that combines a conventional tac promoter and synthetic promoter-ribosome-binding site-terminator (semi-endo PRT) elements in dual plasmids. Additionally, we assessed the antimicrobial activity of violacein against type I methanotrophs. Recombinant E. coli DH5α harboring a hybrid system produced 111.3 ± 19.7 and 0.88 ± 0.23 mg/L of PHB and violacein, respectively, in M9 using xylose as the sole carbon source, without tryptophan supplementation. Using the synthetic PRT system, 528.9 ± 104 mg/g dry cell weight (DCW) of PHB was obtained. Additionally, violacein inhibits the growth of Methylomicrobium alcaliphilum 20Z at 9 µg/mL in nitrate mineral salt medium containing methanol as the sole carbon source. The use of lignocellulose-derived sugars for co-production offers an environmentally sustainable bio-manufacturing approach that contributes to greenhouse gas mitigation and supports the transition toward a circular bioeconomy.
|
|
Scooped by
mhryu@live.com
March 9, 9:41 AM
|
The analysis of ribosome profiling (Ribo-Seq) data has provided evidence that many eukaryotic mRNAs contain translated upstream or downstream ORFs (uORFs/dORFs), but the biological significance of this translation activity remains, for the most part, unknown. One of the principal limitations has been the lack of Ribo-Seq data from several closely related species, precluding the identification of cases in which translation is phylogenetically conserved. Here, by combining Ribo-Seq data from 100 different experiments, we identify 2,332 translated uORFs and 1,008 translated dORFs in S. cerevisiae, which result in microproteins that tend to be highly hydrophobic or positively charged. To study their phylogenetic conservation, we have generated Nanopore direct RNA sequencing data, together with Ribo-Seq data, from six additional Saccharomyces species, spanning an evolutionary period of around 16 million years. We have identified 195 translated S. cerevisiae uORFs that are also translated in other Saccharomyces species; these uORFs are translated at levels comparable to the main coding sequence and display signatures of purifying selection at the level of the encoded microproteins. In contrast, dORFs are translated at very low levels and they are rarely conserved, suggesting much more limited microprotein functionalization. We have also discovered that uORF translation is associated with the formation of alternative transcript isoforms encompassing the region containing the uORFs but not the main protein coding sequence, implying that some microproteins can be produced independently of the main protein product. This work significantly advances our understanding of how initially pervasive uORF translation can result in new microproteins, providing many new candidates for further functional studies.
|

























spytag