Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 6:18 PM
|
The recovery of metagenome-assembled genomes (MAGs) from shotgun metagenomic sequencing is rapidly expanding the availability of representative genomes. However, this practice may skew the representation of specific taxa in real-world datasets. This bias is attributed primarily to the known inefficiencies of sequence-by-synthesis platforms in amplifying GC-rich and AT-rich sequence fragments. Here, we recover 216 medium- and high-quality MAGs from an Australian wetland site. Notably, no MAGs were recovered for some dominant cyanobacterial and proteobacterial species known to be present. A new protocol involving read-based classification and alignment to the MAG dataset demonstrated the highly efficient recovery of low-GC organisms in the Actinobacteria and Bacteroidota phyla. Additionally, the recovery of lost taxonomic information was demonstrated through unmatched sample mapping. The findings suggest a bias towards the recovery of smaller, low-GC organisms in MAG recovery, potentially skewing the global representation of microbial diversity. Our pipeline is made publicly available as a tool to help researchers estimate taxonomic losses following MAG recovery efforts.
|
|
Scooped by
mhryu@live.com
Today, 1:09 PM
|
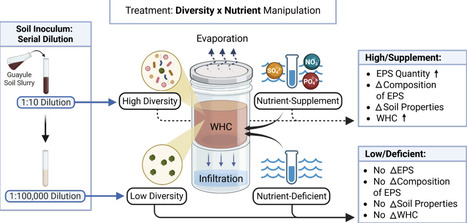
Soil microbial communities have a variety of mechanisms to deal with emerging drought stress. One well-documented mechanism is increased microbial production of extracellular polymeric substances which can potentially change the soil density and water holding capacity. Yet little is known about how microbial diversity influences the functional capacity of EPS formation and the resulting outcomes in water dynamics. To understand more about communal microbiome EPS production, we set up sterile mesocosms where we examined the effects of microbial diversity and nutrient input on these processes. To capture the microhydrology of the mesocosms, we measured water holding, infiltration, evaporation, and soil properties we believe microbes are altering. Our hypothesis stated that if diversity was artificially manipulated, then soil-water properties will be altered via production of EPS. We predicted that low diversity systems would have lower functional diversity, leading to less EPS production, moisture storage, and minimal changes from inert soil media. As predicted, we found that the high-diversity systems had a higher water retention and lower rates of water loss over time than low-diversity systems. This trend was magnified in the nutrient-supplemented treatment, suggesting that EPS production and subsequent water-holding traits are emergent features of the microbiome. Unexpectedly, we observed a correlation between the amount of water retained and the quantity of lipid EPS produced. This suggests that EPS composition, rather than quantity, is determinative of a biofilm's function. In conclusion, it appears that microbial diversity influences soil properties that are important to moisture retention within these systems. To date, the role that microbes and their diversity play in soil hydrology has been severely understudied, so this work aims to build ecological understandings of these systems. These findings are valuable, for if we learn how microbes manipulate soil moisture, we can apply these functions to advance sustainable agricultural practices and enhance ecosystem resilience to water scarcity in arid regions.
|
|
Scooped by
mhryu@live.com
Today, 11:59 AM
|
The development of functional nanomaterials with controlled morphologies is essential for advancements in medicine, electronics and computing, energy, catalysis, and environmental applications. However, conventional synthesis methods often demand high energy input and pose significant environmental challenges. Urease-based biomineralization presents an efficient, eco-friendly alternative for nanomaterial production under mild conditions. In this study, we engineered E. coli to express a urease gene cluster from Sporosarcina pasteurii using CRAGE-Duet technology. The engineered strain successfully synthesized calcium carbonate and calcium phosphate crystals. Expanding the approach, we synthesized metal oxide nanoparticles, including hematite (Fe2O3), and nanocrystalline anatase titanium dioxide (TiO2). These nanomaterials were characterized by electron microscopy, demonstrating the potential of E. coli as a sustainable and versatile platform for green nanomaterial synthesis.
|
|
Scooped by
mhryu@live.com
Today, 11:41 AM
|
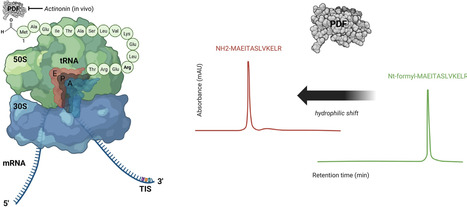
Accurate delineation of bacterial translation initiation sites (TISs) remains a major challenge, as conventional genome annotation and ribosome profiling (Ribo-seq) often lack the resolution to discriminate closely spaced start codons. To overcome these limitations, we developed TRAINSPOTTER (TRAnslation INitiation SPOTTER), a deformylation-assisted N-terminomics workflow that enables direct, proteome-wide detection of nascent N-termini indicative of active translation initiation. TRAINSPOTTER exploits the universal N-terminal formylation of initiator methionine in bacteria: in vitro enzymatic deformylation by peptide deformylase (PDF) generates a diagnostic hydrophilic shift, allowing selective isolation of previously formylated, initiation-derived peptides by COFRADIC-based chromatography. Optional in vivo PDF inhibition transiently enriches formylated N-termini, primarily enhancing detection sensitivity. Integration of pulse SILAC (Stable Isotope Labeling by Amino acids in Cell Culture) labeling confirmed that deformylation-shifted peptides represent newly synthesized N-termini, validating TRAINSPOTTER’s specificity for nascent translation products. Application to E. coli enabled precise mapping of >1000 TIS-indicative N-termini, including numerous alternative and near-cognate start sites, providing direct proteomic evidence for co-expressed N-terminal proteoforms. The method complements and refines Ribo-seq datasets, offering amino acid-level resolution for otherwise ambiguous initiation events. TRAINSPOTTER thus establishes a robust biochemical framework for proteome-wide identification of TISs and advances the experimental annotation of bacterial proteomes.
|
|
Scooped by
mhryu@live.com
Today, 11:33 AM
|
Prokaryotic CRISPR–Cas systems rely on the Cas1–Cas2 protein complex to capture new DNA from mobile genetic elements (MGEs), to form immunological memory that defends against the MGEs. However, the mechanisms by which Cas1–Cas2 locates suitable DNA substrates inside cells remain unclear, limiting our understanding of how CRISPR–Cas immunity arises de novo. We directly visualized functional, DNA-bound Cas1–Cas2 complexes in bacteria, revealing the processes that license Cas1–Cas2 to capture DNA. Visible DNA-bound Cas1–Cas2 complexes formed only when replisomes are actively advancing, accumulating at post-replicative DNA gaps behind replication forks—structures arising during normal genome duplication, which are normally repaired by homologous recombination. Replication stress, which increases replicative DNA gap frequency, enhanced visible Cas1–Cas2 DNA binding. DNA capture by Cas1–Cas2 was strongly stimulated in cells lacking the RecFOR complex, which normally directs DNA gaps to repair. The RecBCD recombination initiator complex was essential for DNA capture by Cas1–Cas2 in these cells. The findings support a model in which naïve CRISPR–Cas adaptation is licensed by abundant replication-dependent DNA repair intermediates, prior to their repair by recombination. This identifies the mechanism co-ordinating Cas1–Cas2 with essential DNA replication and repair processes that all cells need, including when they are hijacked to replicate parasitic MGEs.
|
|
Scooped by
mhryu@live.com
Today, 11:26 AM
|
A suite of artificial-intelligence tools is helping to speed up the process of discovering new antibiotics.
|
|
Scooped by
mhryu@live.com
Today, 11:18 AM
|
Spatial organization within bacterial communities plays a critical role in mediating cell-cell interactions and determining microbial fitness. During infection, Vibrio cholerae undergoes dynamic restructuring of its spatial organization, forming monospecific aggregates thought to enhance survival in the face of the host immune system. However, the effectiveness of its primary weapon against other bacteria—its contact-dependent type VI secretion system (T6SS)—is severely limited in this aggregated form, hampering its ability to compete for space and resources. Here, we show that the presence of competing, co-resident bacteria alters V. cholerae aggregation by modulating toxin co-regulated pilus (TCP) expression through production of interspecies quorum sensing signal autoinducer-2 (AI-2). Using a zebrafish infection model, we found that this quorum sensing-controlled disaggregation enhances the efficacy of T6SS-mediated killing in vivo by promoting intermixing of V. cholerae, thereby increasing cell-cell contact with competitors. This modulation of aggregation has no impact on T6SS activity in vitro, highlighting the context-specific nature of these interactions. We developed a mathematical model to explore these dynamics and observed a fundamental trade-off between potency of V. cholerae T6SS and its sensitivity to the presence of competing bacterial species. Our findings reveal a core mechanism underlying V. cholerae colonization wherein it uses quorum sensing to dynamically balance between protective aggregation to survive host defenses and dispersed infiltrative intermixing to facilitate elimination of competitors. The pathogenic bacterium Vibrio cholerae forms aggregates within its host, which helps resisting host defenses. Here, Virgo et al. show that, upon sensing signals produced by competing bacteria, V. cholerae disperses its aggregates, facilitating interspecies mixing and the killing of the competing bacteria via the pathogen’s contact-dependent secretion system.
|
|
Scooped by
mhryu@live.com
Today, 9:57 AM
|
Constructing multi-step bioinformatics workflows, from read quality control through genome assembly to functional annotation, requires expertise in both biology and computational tool selection, creating a bottleneck for scalable and reproducible analysis. We present the KBase Research Agent, a multi-agent system for automating such workflows within the DOE Systems Biology Knowledgebase (KBase). Given a set of sequencing reads and a research objective, the agent constructs an analysis plan grounded in KBase documentation and a Knowledge Graph (KG) of the KBase application catalog, then selects, parameterizes, validates and executes appropriate KBase applications to carry out the workflow. The resulting analysis is preserved as a reproducible KBase Narrative. We evaluate the system's planning and execution quality against ground truth constructed from reference workflows derived from peer-reviewed Microbiology Resource Announcements. We further apply the agent to 100 previously unanalyzed bacterial isolate genomes from the JGI IMG/M database, where it autonomously performed read quality control, genome assembly, taxonomic classification with GTDB-Tk, and downstream analysis producing annotated genomes, reproducible Narratives, and draft manuscripts without human intervention. Across these experiments, the KBase Research Agent demonstrates the feasibility of domain-grounded, end-to-end scientific workflow automation in a production bioinformatics platform.
|
|
Scooped by
mhryu@live.com
Today, 9:41 AM
|
Endophytic fungi residing within medicinal plants represent a rich and sustainable source of bioactive secondary metabolites with diverse pharmacological applications. These symbiotic microorganisms establish mutualistic relationships with their host plants, contributing to enhanced stress tolerance, growth promotion, and defense against pathogens. In recent years, endophytic fungi have gained considerable attention as alternative biofactories for the production of valuable compounds such as alkaloids, terpenoids, phenolics, lignans, and polysaccharides. These metabolites exhibit a wide range of biological activities, including antimicrobial, antiviral, anticancer, antioxidant, anti-inflammatory, and antidiabetic effects. Advances in cultivation strategies, such as OSMAC, co-culture, and epigenetic modification, have significantly improved metabolite yield and diversity. Moreover, the integration of genomics, transcriptomics, and metabolomics has revolutionized the understanding of biosynthetic gene clusters and metabolic pathways, enabling the discovery of novel compounds and optimization of production processes. Despite these advances, challenges such as low yield, silent gene clusters, and difficulties in large-scale production remain significant barriers. Nevertheless, continued progress in multi-omics technologies, synthetic biology, and biotechnological tools holds great promise for unlocking the full potential of endophytic fungi. Herein, endophytic fungi represent a powerful and eco-friendly platform for the development of new therapeutic agents and sustainable pharmaceutical applications.
|
|
Scooped by
mhryu@live.com
Today, 9:32 AM
|
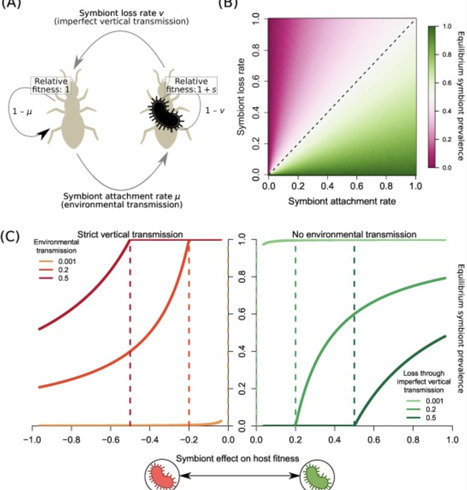
Host-associated bacteria are found across the tree of life. In this opinion article, we propose that population genetics theory can be used to probe the conditions that form the path toward such symbioses. We illustrate how mutation-selection models generate insights into the maintenance of a symbiont under transmission between generations and from the environment. We outline how basic features of host population size and life history shape the fixation of a heritable symbiont in a host population, suggesting elevated fixation probabilities in long-lived hosts. Finally, whenever the fitness effects of the symbiont vary over time, reduced efficiency of selection increases the fixation of a deleterious symbiont. Our predictions of the properties of hosts, symbionts, and their ecological contexts that impact symbiont establishment frame expectations across systems.
|
|
Scooped by
mhryu@live.com
June 7, 12:07 PM
|
Life originated in the absence of oxygen. Despite its substantial energetic advantages, many modern microbes remain obligate anaerobes, confined to anoxic niches such as the mammalian gut. Why these organisms cannot tolerate oxygen has remained unresolved for more than two centuries. Here, using integrated multi-omics analyses, we identify a network of interlocking vulnerabilities in central metabolism, biosynthetic pathways, and redox homeostasis that together impose an aerobic growth barrier in the obligate anaerobic commensal Bacteroides thetaiotaomicron. Rational repair of these vulnerabilities restores metabolic integrity and progressively enhances oxygen tolerance, yielding engineered strains capable of robust growth at 10% O2 and markedly improved resilience in the oxygenated, inflamed gut. These findings define a molecular basis for obligate anaerobiosis and establish a framework for engineering commensal bacteria to function in oxidative environments, expanding their ecological range and therapeutic potential.
|
|
Scooped by
mhryu@live.com
June 7, 11:50 AM
|
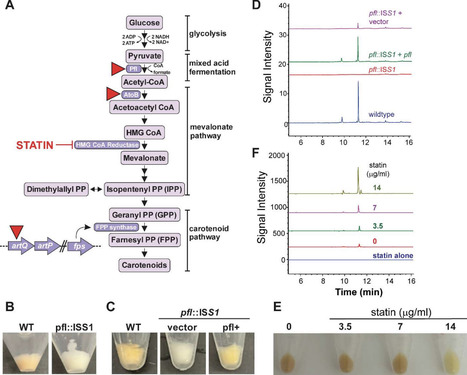
Streptococcus pyogenes is a major human-restricted pathogen capable of both localized and systemic diseases, as well as causing post-infectious acute and chronic rheumatological conditions. Despite its well-documented clinical importance and over a century of extensive research, gaps persist in understanding this pathogen. We report the discovery of a novel pigment produced by S. pyogenes when cultured in a replete, chemically defined medium. Color development accumulates during growth, requires exposure to oxygen, and remains associated with the bacterial cell. Though only 20% of a small strain collection produced the pigment (8 of 40), positive cultures were overrepresented by M1 and M89 serotypes. Given that pigments are critical virulence and fitness determinants in pathogens like Staphylococcus aureus and Streptococcus agalactiae, here we describe initial attempts to characterize S. pyogenes pigment biosynthesis, regulation, and potential benefits to the organism. 20,405 transposon mutants were screened for pigment loss in liquid culture, and we identified 94 independent hits enriched in pathways associated with isoprenoid biosynthesis, purine biosynthesis, guanosine transport, and mixed acid fermentation. Although color development requires oxygen, the extracted pigment did not provide antioxidative activity as compared to non-pigmented extracts. Pigment production was inhibited when the Rgg2/Rgg3 quorum-sensing system was active, though by unknown means. Treatment of RAW-Blue macrophages with the extracted pigment significantly reduced NFκB activation, suggesting a potential anti-inflammatory effect. Overall, this study describes a previously uncharacterized pigment produced by Streptococcus pyogenes and provides insights into its oxygen-dependent production and associated metabolic pathways.
|
|
Scooped by
mhryu@live.com
June 7, 11:42 AM
|
Partner-specific protein-protein binding site prediction, identifying which residues of a protein form the interface when bound to a specific partner, remains a challenging task with significant implications for drug discovery and understanding of protein structure and function. Existing computational methods are limited by small training datasets, inconsistent redundancy filtering, and reliance on three-dimensional structural information at test time. Here we present a sequence-only, partner-specific protein-protein interface predictor called Handshake. It combines ProstT5, a protein language model pre-trained on structural data, with Low-Rank Adaptation (LoRA), a cross-chain attention mechanism and a contact supervision head. Our method can detect both binding interfaces and pairwise contact matrices. We trained our model on very large datasets of non-redundant protein-protein pairs derived from the PPInterface dataset, the most comprehensive structural protein-protein database to date, and evaluated it on systematically filtered benchmarks at four redundancy thresholds (30%--90% sequence identity). We demonstrate that sequence redundancy inflates reported AUROC by up to 0.079 and MCC by up to 0.145 on identical models, representing a substantial methodological confound in the field. Even at 30% redundancy threshold, our results (AUROC=0.811, MCC=0.367, F1=0.45) exceed the best published sequence-only result on this convention. Our method also achieves comparable performance to existing partner-specific methods that use explicit structural information. The comprehensive training and evaluation dataset, in addition to the systematic redundancy inflation, can help gain insight into protein-protein interactions and the abilities and limitations of current detection methods.
|
|
|
Scooped by
mhryu@live.com
Today, 6:09 PM
|
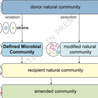
Consortia of microbial isolates, also known as synthetic communities (SynComs), are increasingly used to study and harness microbe-microbe and microbe-host interactions. Since “synthetic” potentially evokes negative connotations, we propose adopting the term “Defined Microbial Community” for practical applications.
|
|
Scooped by
mhryu@live.com
Today, 12:00 PM
|
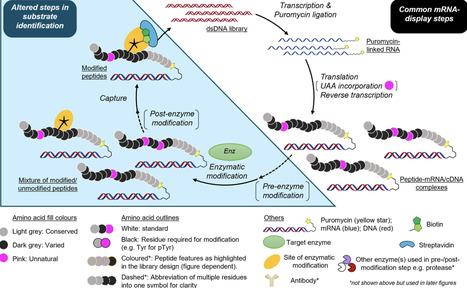
Among display technologies, mRNA display has emerged as a powerful approach to identify de novo ligands for proteins of interest. Applying the same methods to studying the substrates of protein/peptide-modifying enzymes has received much less attention, but progress in this area has accelerated rapidly over the last 5 years. In this article, we review the published literature to date (up to December 2025) of dedicated efforts to identify and understand enzyme substrate preferences using mRNA- or cDNA-display. We include observations of trends from these reports over time and reflections on where the area may go in the future. We hope this will serve as a useful primer for researchers in this and related areas.
|
|
Scooped by
mhryu@live.com
Today, 11:55 AM
|
The large serine recombinase Bxb1 catalyzes recombination between DNA molecules containing compatible attP and attB sequences, offering broad applications in genome engineering and gene therapies. Here, we present cryo-electron microscopy structures of the Bxb1-attP-attB synaptic complex in four distinct functional states during its recombination cycle. Notably, the Bxb1 complex structures in the pre-, mid-, and post-strand-exchange states explain how the attP- and attB-bound Bxb1 dimers are assembled into a tetrameric synaptic complex and how an approximately 180° rotation occurs between the left and right dimers after DNA cleavage, thereby enabling DNA strand exchange and religation. Furthermore, we engineered Bxb1 variants with altered DNA preferences and enhanced recombination activity, which improved programmable gene integration in human cells. Overall, our findings advance the mechanistic understanding of large serine recombinases and provide a structural framework for future engineering of Bxb1-mediated genome integration technologies.
|
|
Scooped by
mhryu@live.com
Today, 11:37 AM
|
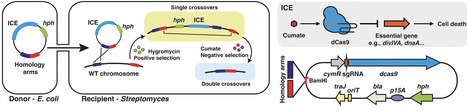
Streptomycetes are prolific producers of bioactive natural products, but many of the biosynthetic gene clusters (BGCs) are silent in the laboratory. Genetic manipulation is important to unlock their full potential. CRISPR–Cas-based genome editing has greatly advanced genetic engineering in Streptomyces. However, several challenges remain, including Cas nuclease toxicity, unintended genomic rearrangements, and elimination of the delivery plasmid. Here, we present a novel genome editing strategy that harnesses cumate-inducible CRISPR interference (CRISPRi) to transiently knockdown essential genes such as divIVA or dnaA as counterselectable marker. This enforces loss of the vector backbone, promotes homologous recombination, and yields markerless mutants by loss of the antibiotic resistance cassette during the final recombination step. We demonstrate the versatility of the ICE system (Inducible CRISPRi targeting an Essential gene) by (i) deleting four BGCs in Streptomyces coelicolor M145, (ii) inserting both a promoter and a large BGC, and (iii) introducing precise single-nucleotide substitutions. Furthermore, deletion of the prodigiosin BGC elicited expression of a poorly expressed BGC for prolinolexin lipopeptides in Streptomyces roseifaciens DSM 106196T. Considering that different essential genes may be targeted, we anticipate that inducible CRISPRi-based counterselection may be adaptable to genome editing strategies in a broad range of microbial systems.
|
|
Scooped by
mhryu@live.com
Today, 11:28 AM
|
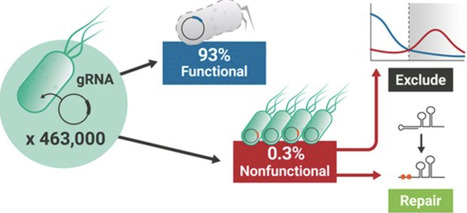
The Cas9 nuclease has become central to modern methods and technologies in synthetic biology, largely due to the ease with which it can be targeted to specific DNA loci via guide RNAs (gRNAs). Reports vary widely on the actual specificity of this targeting, with some studies observing 60% of gRNAs possessing no activity against the genome, yet an assumption persists within the E. coli community that inactive gRNAs are rare. To resolve these contradictions, we evaluated the activity of 463 000 unique gRNAs in the E. coli K12 MG1655 genome. We show that the overwhelming majority (at least 93%) of unique gRNAs are functional while only 0.3% are nonfunctional. These nonfunctional gRNAs exhibit strong spacer self-interaction, which can either be excluded using a simple design rule or “repaired” during library design. Finally, this work provides the greater microbial synthetic biology community both a set of nearly half a million empirically evaluated E. coli gRNAs as well as a thoroughly evaluated experimental procedure, complete with appropriate controls for Cas9 activity, for conducting Cas9 assays in E. coli specifically and bacteria more generally. Lastly, we have produced a webapp to allow users to easily browse and extract gRNA sequences from the E. coli genome, which can be accessed at https://grna.ornl.gov.
|
|
Scooped by
mhryu@live.com
Today, 11:20 AM
|
Base editing shows great potential in research and clinical applications. Current iterations of the deaminases used to create precise single-nucleotide changes via base editing exhibit undesirable effects, including off-targeting, off-base editing, and bystander editing. Current deaminases are derived from either larger eukaryotic deaminases, which exhibit high levels of Cas-independent DNA targeting, or from evolved variants of the smaller E. coli TadA protein (ecTadA), which exhibits off-base editing. To overcome the limitations inherent to using a single protein sequence for engineering, we diversified newly identified TadA orthologs by DNA shuffling to yield millions of training sequences for measuring base editor efficiency. We trained generative models on the performance data from the pools of variants and drew on information-theoretic insights to efficiently explore the sequence space to generate diverse and high-performing deaminases. From a single round of diversification, we created a small set of novel and specific cytosine and adenosine deaminases that were markedly distinct in sequence from published base editor deaminases. We found that our model-created deaminases generally outperform those we identified through typical directed evolution. The novel compact deaminases identified here show high on-base activity, comparable to the leading published base editors, and with demonstrably lower off-base activity.
|
|
Scooped by
mhryu@live.com
Today, 11:10 AM
|
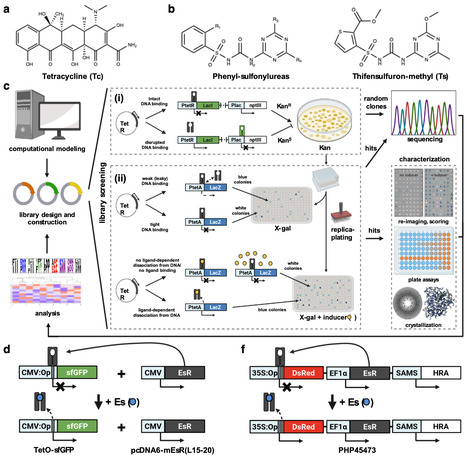
Chemically inducible expression systems enable transgene expression regulation in response to external small molecules. Tetracycline repressor (TetR)-based gene switches work in plants, but antibiotics are neither approved nor advisable for crop use. Here we report engineering of TetR mutants that respond to approved sulfonylurea (SU) herbicides instead of antibiotics. Designed variants show low-nanomolar EC50 values for ethametsulfuron-methyl (Es) or chlorsulfuron and tightly bind the Tet operator sequence, but only in the absence of corresponding SUs. Crystal structures of two repressors in complex with their respective SU ligands reveal extensive interactions explaining their strong binding. The Es repressor-based gene switch is introduced into tobacco, soybean, maize, rice, and Arabidopsis, and robust reporter gene activation is observed upon herbicide application. Addition of a repressor-regulated siRNA targeting the repressor transcript increases the magnitude and spatial distribution of the response following herbicide treatment and results in a partially bistable gene switch. The SU repressors also function well in mammalian cell culture and may enable regulation of additional genes in conjunction with TetR. Practical chemically inducible expression systems for crop plants remain lacking. Here, the authors report the design and evolution of the tetracycline repressor into sulfonylurea herbicide-responsive gene switches and show their application in multiple crops as well as mammalian cell.
|
|
Scooped by
mhryu@live.com
Today, 9:55 AM
|
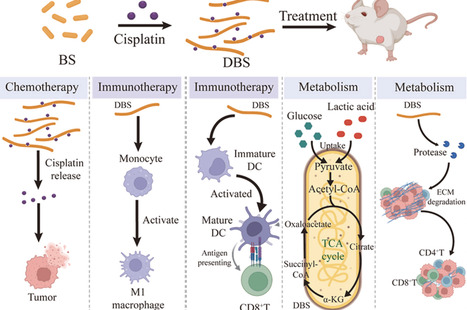
Cancer treatment remains a critical global health challenge, which often requires multimodal and targeted therapeutic strategies, yet integrating these modalities typically demands complex formulations. Oncolytic bacterial therapy offers unique opportunities for targeted drug delivery and immune modulation, yet clinical translation has been hindered by safety concerns and tedious engineering modification. Here, we report a deformable Bacillus subtilis (DBS), constructed through one-step cisplatin-induced morphological transformation that elongates the bacterium while simultaneously incorporating chemotherapeutic payloads. The resulting DBS integrated three synergistic antitumor mechanisms: Chemotherapy, Starvation, and Immunotherapy. Metabolically, DBS actively consumes glucose and lactate within the tumor microenvironment (TME), thereby depriving tumor cells of key nutrients and reshaping the microenvironment to become more permissive to therapy. Concurrently, anaerobe-targeting DBS releases the encapsulated cisplatin locally in the TME, enabling direct cytotoxic killing while limiting systemic exposure. Furthermore, DBS leverages its intrinsic immunogenicity to potentiate immune cell activation, while its elongated morphology prolongs tumor retention and facilitates enhanced interactions with infiltrating immune cells. Proteases secreted by DBS hydrolyze the extracellular matrix, facilitating immune cell infiltration and boosting anti-tumor immune responses. Surprisingly, DBS exhibits intestinal homing and is safely cleared through fecal excretion after treatment, minimizing systemic side effects. This study presents an efficient and safe live-bacterial platform for cancer therapy, underscoring the broad potential of bacterial morphology engineering as a facile strategy to integrate multiple synergistic antitumor mechanisms within a single microbial therapeutic.
|
|
Scooped by
mhryu@live.com
Today, 9:37 AM
|
Predicting protein function is a fundamental and challenging task that requires integrating diverse biological data modalities to capture complex functional relationships. Traditional machine learning methods often rely on single modalities or combine only a limited number (typically two), without aligning them in a unified representation, thereby constraining predictive accuracy. Moreover, most existing machine learning approaches are limited to preselected subsets of Gene Ontology (GO) function terms with sufficient annotations, making the prediction of novel function terms a persistent challenge. Here, we present FunBind, a multimodal AI model that jointly learns from five modalities, i.e., protein sequences, textual descriptions, domain annotations, structures, and GO terms, to enhance prediction accuracy and infer previously unseen functions. FunBind operates in two modes: (1) self-supervised pretraining using contrastive learning to align the sequence modality with other heterogeneous modalities in a unified latent space, enabling unsupervised zero-shot function prediction, and (2) supervised fine-tuning of the pretrained model to leverage all non-function modalities for comprehensive and accurate function classification. Our results show that FunBind’s zero-shot capabilities allow it to generalize effectively to novel function terms never encountered before, while its joint multimodal fine-tuning strategy outperforms single-modality models and current state-of-the-art deep learning methods in typical function prediction settings.
|
|
Scooped by
mhryu@live.com
June 7, 12:23 PM
|
Plant metabolism underpins the food, fiber, and fuel that support our economy, driving strong interest in new strategies to rewire plant metabolism for emerging applications. While most synthetic biology efforts are reliant on genetic engineering, plants can be manipulated in many other ways that remain comparatively underexplored. Across nature, diverse organisms, including bacteria, fungi, and insects, have evolved sophisticated mechanisms to exploit plant metabolic richness, reshaping it for purposes that span from basic nutrition to the construction of complex, novel structures for shelters. These interspecies interactions and non-model systems represent unique manners in which plants can be reprogrammed or hijacked by other organisms, offering inspiration for novel approaches to engineering plant metabolism. By better understanding the basis of how organisms induce these remarkable transformations in plants, we can expand the conceptual boundaries of synthetic biology and reveal alternative routes to manipulating plants for the production of a diverse array of valuable compounds and materials. Deeper insight into these mechanisms will yield novel blueprints for rethinking the scope and breadth in which we can redesign plant metabolism across many applications.
|
|
Scooped by
mhryu@live.com
June 7, 11:53 AM
|
Biotransformation of synthetic steroid drugs by gut and environmental bacteria shapes the activity within the host, persistence, and environmental fate of these widely used pharmaceuticals. However, the scope of bacterial transformations of synthetic steroids, the enzymes involved, and the role of microbial cooperative metabolism in these processes remain poorly understood. Here, we systematically investigated the biotransformation of 20 synthetic and 2 natural steroids, including clinically relevant estrogens, progestogens, corticosteroids, and prodrugs, across 8 intestinal and 4 environmental bacterial species. We identified more than 130 biotransformation products and found that the tested bacteria catalyzed diverse reactions including ester hydrolysis, oxidation-reduction chemistry, and steroid side-chain cleavage. Notably, the aerobic environmental bacterium Sphingobium herbicidovorans catalyzed desmolase-like steroid side-chain cleavage, a transformation not previously reported in aerobic bacteria. Combining homology searches, gain-of-function screening, and expression proteomics we identified six steroid-transforming enzymes in S. herbicidovorans. We further demonstrated that distinct bacterial species cooperatively metabolize synthetic steroids through interspecies metabolic cross-feeding, enabling sequential activation and metabolism of corticosteroids across microbial communities. Together, our findings uncover previously unrecognized bacterial enzymes and community-level interactions involved in synthetic steroid metabolism, directly linking metabolites, enzymes, and community interactions and establishing microbial biotransformation as an important determinant of steroid drug fate in host-associated and environmental ecosystems, where mechanistic resolution of bacterial steroid pathways is essential to predict metabolic interactions within and across microbial communities.
|
|
Scooped by
mhryu@live.com
June 7, 11:44 AM
|
Current pangenome construction methods rely largely on nucleotide or protein sequence alignment, limiting their ability to detect remote orthologs and semantic relations. We introduce a novel method that leverages protein language model embeddings to capture functional and semantic relationships beyond sequence similarity. Our approach employs approximate nearest-neighbor search coupled with a clustering step utilizing HDBSCAN, DBSCAN, or weighted single-linkage clustering with multiple similarity thresholds. The method utilizes GPU acceleration, dynamic batching, and ONNX optimization to scale approximately linearly with the number of proteins, enabling the analysis of datasets containing millions of proteins. We evaluated our approach on a randomly sampled subset of OrthoDB and the CAFA5 dataset, benchmarking it against SCARAP. SCARAP is a recently published tool with similar performance to a variety of other common tools for computing pangenomics. Our benchmarking demonstrates that our method produces more specific clusters than SCARAP across both datasets. SCARAP excelled in term consistency within clusters on the OrthoDB dataset, where labels are inferred with sequence alignment (using MMseqs2). Both methods face a significant degradation in term consistency when transitioning to the experimentally validated CAFA5 dataset, ultimately resulting in similar term consistency scores for both approaches. Crucially, our approach yields superior cluster quality on both datasets and significantly outperforms SCARAP across all metrics of functional consistency and coherence on the experimental CAFA5 dataset. Finally, we demonstrate the method's scalability and utility by characterizing the pangenome of 1,034 Streptomyces genomes. The pipeline is available for use at our GitHub: https://github.com/jakob949/pan_genome
|