 Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 4:02 PM
|
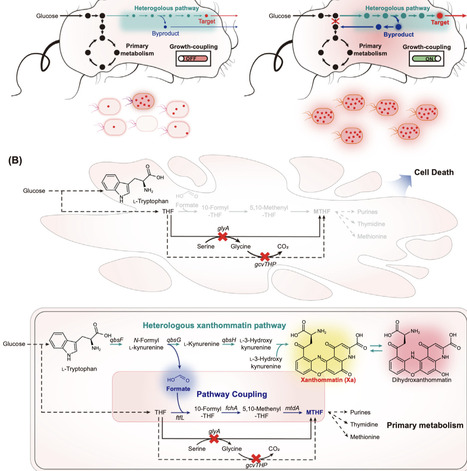
Microbial natural product biosynthesis often suffers from metabolic burden and genetic instability. Bushin et al. introduce a synthetic C1 feedback loop that couples xanthommatin production to cell survival, enabling adaptive laboratory evolution to achieve gram-scale production. This strategy offers a generalizable route to engineer cell factories for complex natural products.
|
|
Scooped by
mhryu@live.com
Today, 3:45 PM
|
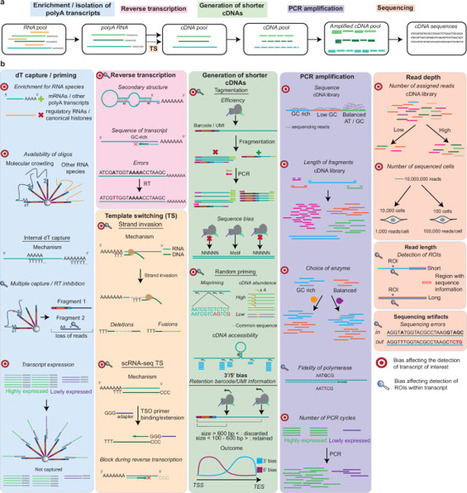
Current single-cell RNA sequencing (scRNA-seq) methods suffer from biases that restrict the detection of cellular transcripts to 10–40% of total RNAs. This hinders the identification of transcripts of interest. Additionally, information retrieved from most high-throughput scRNA-seq methods is confined to untranslated regions toward transcript ends, resulting in loss of detail in internal regions. In this review, we outline biases in scRNA-seq protocol steps that limit transcript and region detection. We then discuss the advantages and disadvantages of targeted sequencing solutions, grouped into five categories according to the protocol step they target. Finally, we present a decision tree that guides researchers in selecting the most suitable targeted method for their experiment. A review of targeted single-cell RNA sequencing methods provides a practical guide to help researchers select the method most suited for their own experiments.
|
|
Scooped by
mhryu@live.com
Today, 3:39 PM
|
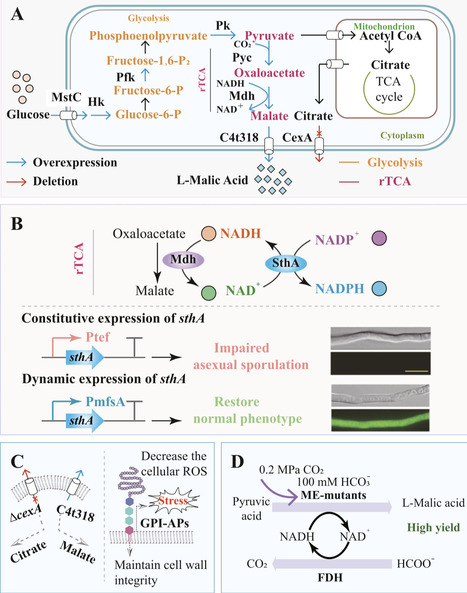
L-Malic acid, a versatile platform chemical, is increasingly manufactured through sustainable microbial fermentation. The filamentous fungus Aspergillus niger stands as a premier cell factory for its industrial production due to its native production capacity and metabolic versatility. This review summarizes cutting-edge advances in elucidating and engineering the L-malic acid biosynthetic pathway in A. niger. A core focus lies on systems metabolic engineering and critical enabling tools that collaboratively redirect carbon flux toward enhanced L-malic acid synthesis. We further discuss enduring challenges in industrial-scale-up, including process intensification and downstream processing, and outline promising future directions that integrate systems biology with innovative bioprocess technologies to advance the economic viability of microbial L-malic acid production.
|
|
Scooped by
mhryu@live.com
Today, 3:27 PM
|
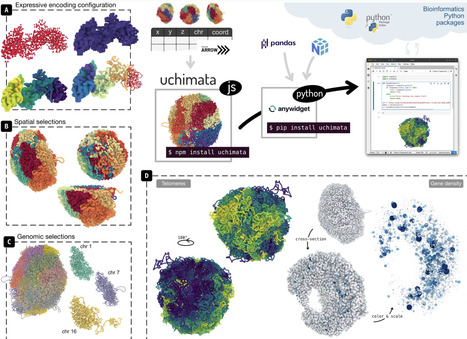
Uchimata is a toolkit for visualization of 3D structures of genomes. It consists of two packages: a Javascript library facilitating the rendering of 3D models of genomes, and a Python widget for visualization in Jupyter Notebooks. Main features include an expressive way to specify visual encodings, and filtering of 3D genome structures based on genomic semantics and spatial aspects. Uchimata is designed to be highly integratable with biological tooling available in Python.
|
|
Scooped by
mhryu@live.com
Today, 3:18 PM
|
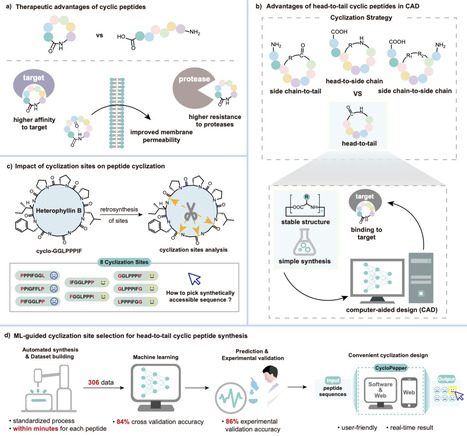
Cyclic peptides exhibit remarkable stability, membrane permeability, and binding affinity, positioning them as promising therapeutics. However, their synthesis, particularly on-resin head-to-tail cyclization, remains challenging, with cyclization site selection critically influencing yield. Here, we introduce a machine learning (ML) approach to predict cyclization outcomes, leveraging CycloBot, our fully automated cyclic peptide synthesis platform. Using this system, we generate a standardized dataset of 306 cyclic peptides (2–14 residues) and develop an ML model achieving an average prediction accuracy of 84%. Experimental validation with 74 random and therapeutic peptides showed an 86% prediction consistency. To facilitate practical use, we built CycloPepper, a user-friendly platform available through both web and software interfaces, enabling rapid cyclization site assessment. This tool effectively identified potential cyclization sites for disease-targeting peptides, including cancer biomarkers. Our work illustrates the potential of ML-assisted synthesis to streamline cyclic peptide synthesis and accelerate therapeutic discovery. Cyclic peptides are promising therapeutics, but their synthesis is often inefficient and sequence-dependent. Here, the authors present CycloPepper, a machine learning–guided platform that predicts cyclization outcomes and enables automated synthesis to accelerate cyclic peptide drug development.
|
|
Scooped by
mhryu@live.com
Today, 2:28 PM
|
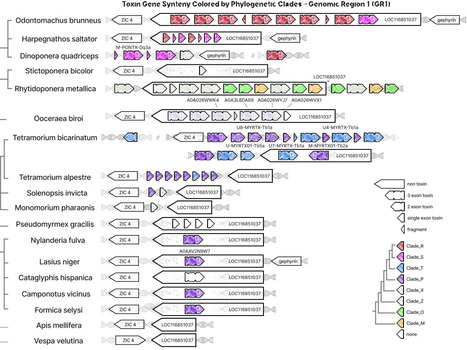
Venoms from ant (Formicidae) are chemically extraordinarily diverse, yet their genomic architecture and evolutionary dynamics remain opaque. The "aculeatoxin hypothesis" assumes that most venom peptides in stinging insects (aculeate) evolutionarily derive from a single gene family, the aculeatoxins. The recent refutation of this hypothesis for bees raises fundamental questions about venom evolution in ants, which contribute most aculeatoxins. To trace venom peptide evolution, we performed synteny-aware comparative genomics across 25 ant species spanning major subfamilies. We analyzed phylogeny through sequences, structures, and embeddings from protein Language Model (pLMs). We identified three conserved genomic regions (GR1-GR3) as evolutionary hotspots for ant venom genes, each exhibiting distinct evolutionary dynamics. Most remarkably, we discovered genuine melittin orthologs in ants at the conserved bee syntenic position (GR2), pushing the origin of this scaffold back to early aculeates, with a possible origin deeper in Hymenoptera. Gene copy numbers vary dramatically (0-17 genes per region), with predatory species showing expansions and formicine ants (subfamily Formicinae, which rely on formic acid spraying) showing reductions. Twenty-two distinct toxin clades emerge, with region-specific distributions suggesting repeated recruitment to conserved platforms. Ants evolutionarily succeed by combining single-copy conservation (bee-like at GR2), massive gene duplication (snake-like at GR1), and repeated lineage-specific recruitment to conserved genomic platforms (GR3). This multi-modal evolution on stable genomic scaffolds, evidenced here, reconciles previously conflicting models of venom evolution and reveals how genomic architecture constrains, yet repeatedly enables, molecular innovation. Our findings highlight a general principle of genome evolution: complex adaptive traits can arise not from a single origin or mechanism, but through recurrent reuse of permissive genomic loci shaped by ecology. Such principles are likely relevant not only to other venomous animals, but more broadly to the evolution of complex multi-genic traits.
|
|
Scooped by
mhryu@live.com
Today, 2:03 PM
|
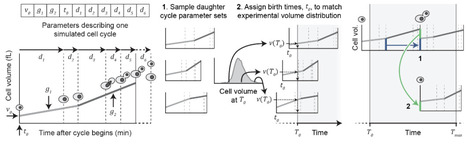
The cell division cycle is characterized by oscillatory dynamics in regulatory mechanisms and biosynthesis, coordinated with genome replication and segregation. To understand these dynamics, quantitative cell cycle-dependent protein concentration data is essential. Unfortunately, accurate resolution of cell cycle-dependent protein dynamics is challenging because single-cell proteomics is currently infeasible and bulk proteomics requires inherently imperfect cell synchronisation. Here, we developed a computational method to deconvolve cell cycle-dependent protein concentration dynamics and applied it to new budding yeast bulk proteome data. Key to this method was a yeast population model, parameterised with experimental cell cycle progression and volume growth data, for quantifying the desynchronisation in sampled populations. We performed deconvolution on 3373 proteins, using cross-validation to determine regularisation parameters, and identified 563 proteins with cell cycle-dependent dynamics. Many of these dynamics were consistent with known yeast biology and dynamic proteins were enriched for several metabolic process, extending previous observations and supporting the emerging picture of metabolic activity as varying substantially over cell cycle phases. We consider the generated cell cycle-resolved budding yeast proteome data a key resource.
|
|
Scooped by
mhryu@live.com
Today, 1:29 PM
|
Designing synthetic biomolecular condensates, or membraneless organelles, offers insights into the functions of their natural counterparts and is equally valuable for cellular and metabolic engineering. Choosing E. coli for its biotechnological relevance, we deploy RNA nano-technology to design and express non-natural membraneless organelles in vivo. The designer condensates assemble co-transcriptionally from branched RNA motifs interacting via base-pairing. Exploiting binding selectivity, we express orthogonal, non-mixing condensates, and by embedding a protein-binding aptamer, we achieve selective protein recruitment. Condensates can be made to dissolve and reassemble upon thermal cycling, thereby reversibly releasing and re-capturing protein clients. The synthetic organelles are expressed robustly across the cell population and remain stable despite enzymatic RNA processing. Compared with existing solutions based on peptide building blocks or repetitive RNA sequences, these nanostructured RNA motifs enable algorithmic control over interactions, affinity for clients, and condensate microstructure, opening further directions in synthetic biology and biotechnology. Designing synthetic biomolecular condensates offers biological insights and is valuable for cellular and metabolic engineering. Here, the authors express orthogonal, unnatural condensates with protein binding aptamers in E. coli, which can dissolve and reassemble upon thermal cycling.
|
|
Scooped by
mhryu@live.com
Today, 1:14 PM
|
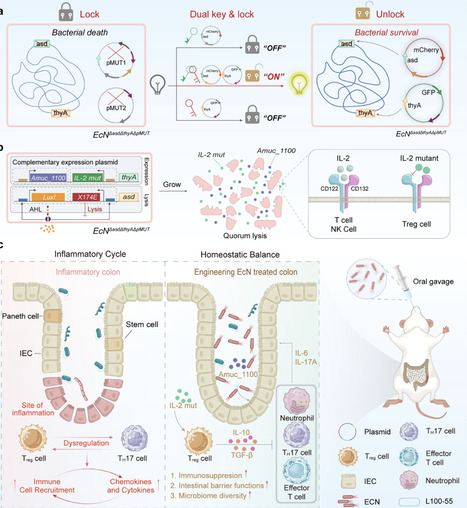
Engineered bacteria offer a promising therapeutic platform but often display plasmid instability and antibiotic dependence. A synthetic dual-key genetic circuit is established in E. coli Nissle 1917 (EcN) by deleting both asd and thyA genes, generating an auxotrophic chassis that requires dual-plasmid complementation for survival. The “lysis module” restores asd function and incorporates a quorum-sensing system for self-regulated lysis and controlled protein release (“One Key”). By contrast, the “expression module” complements thyA and co-delivers an interleukin-2 (IL-2) mutant and the membrane protein Amuc_1100 to modulate immune balance and repair the intestinal barrier (“Dual Keys”). This dual-key design enabled antibiotic-free plasmid stability, precise population control, and sustained therapeutic protein secretion. Oral administration of the engineered strain significantly alleviated colitis in mice by enhancing regulatory T-cell expansion, restoring epithelial integrity, and reshaping the gut microbiota. This modular system provides a safe, stable, and programmable strategy for live bacterial therapy against immune and mucosal diseases.
|
|
Scooped by
mhryu@live.com
Today, 12:24 PM
|
Accounting for 12% of global solid waste, Poly (ethylene terephthalate) (PET) is one of the most abundantly produced synthetic polymers. While PET offers substantial commercial benefits, its widespread use has led to disproportionate environmental hazards due to its resistance to degradation. To address this problem, several solutions have been proposed, including enzymatic degradation via PETase, MHETase, and Cutinase. Among these, PETase exhibited significant PET-degrading activity. However, the application of PETase has been hampered by its lack of robustness to pH, temperature ranges, and slow reaction rates. Hence, it has become novel enzymes that can overcome these limitations and function efficiently. In this study, we utilized an integrated in silico bioinformatics pipeline to identify and characterise novel PETase candidates from the Thermophilic actinobacteria Thermobifida cellulosilytica and Thermobifida halotolerans species. The PlasticDB database contains 228 plastic-degrading enzyme sequences. In which PETase (00188) is significantly homologous with two putative proteins, Hydrolase (ALF00495.1) and hypothetical protein (WOZ56011.1). The discrete optimized protein energy (DOPE) scores, stereochemical assessments, and homology modeling results closely mirrored our findings for both proteins, supporting their structural stability. The molecular dynamics simulations revealed that the putative ALF00495 variant exhibited more extensive and robust hydrogen-bonding networks, enhanced conformational stability, and increased structural compactness compared to the reference enzyme. The present in silico investigation underscores the potential of putative ALF00495 as a highly effective PETase biocatalyst for polyethylene terephthalate (PET) degradation. Collectively, these findings illustrate the utility of computational approaches of novel PET-degrading enzymes, thereby facilitating the development of sustainable biotechnological strategies to mitigate global plastic pollution.
|
|
Scooped by
mhryu@live.com
Today, 12:19 PM
|
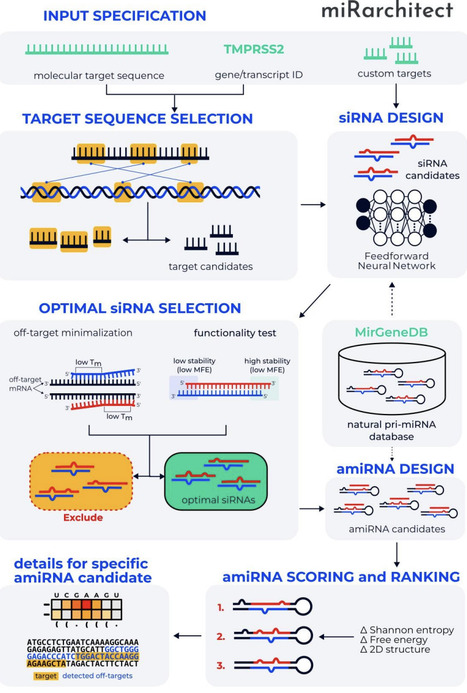
Artificial microRNAs (amiRNAs) offer a powerful strategy for targeted gene silencing, but their rational design is limited by complex sequence-structure-processing relationships and the lack of tools capable of optimizing efficacy and specificity. To address this need, we developed miRarchitect, a web-based platform that uses machine learning to support the customizable design of amiRNAs. miRarchitect integrates neural network-guided target-site selection, siRNA insert design, and scaffold choice, utilizing large-scale data from human primary microRNAs (pri-miRNAs) and next-generation sequencing. The platform generates molecules that closely resemble endogenous pri-miRNAs and includes comprehensive off-target analysis to enhance specificity. Experimental validation targeting TMPRSS2 and ACE-2 confirmed precise processing, robust knockdown, and high specificity of miRarchitect-designed amiRNAs. In comparative benchmarking, miRarchitect consistently produced functional amiRNAs, whereas only half of the top candidates generated by other tools showed measurable activity. miRarchitect is freely available at https://rnadrug.ichb.pl/mirarchitect and provides an intuitive interface with an automated workflow for generating, ranking, and selecting candidate amiRNAs for research and therapeutic applications.
|
|
Scooped by
mhryu@live.com
Today, 12:14 PM
|
Optimizing synonymous codon sequences to improve translation efficiency, RNA stability, and compositional properties is challenging because the search space grows exponentially with protein length and objectives interact through long range RNA structure. Dynamic programming-based methods can provide strong solutions for fixed objective combinations but are difficult to extend to additional constraints. Deep generative models require large-scale, high-quality mRNA sequence datasets for training, limiting applicability when such data are scarce. Reinforcement learning naturally handles sequential decision-making but faces challenges in codon optimization due to delayed rewards, large action spaces, and expensive structural evaluation. We present CodonRL, a reinforcement learning framework that learns a structural prior for mRNA design from efficient folding feedback and demonstration-guided replay, and then enables user-controlled multi-objective trade-offs during inference. CodonRL uses LinearFold for fast intermediate reward computation during training and ViennaRNA for final evaluation, warms up learning with expert sequences to accelerate convergence for global structure objectives, and introduces milestone-based intermediate rewards to address delayed feedback in long range optimization. On a benchmark of 55 human proteins, CodonRL outperforms GEMORNA, a state-of-the-art codon optimization method, across multiple metrics, achieving 9.5% higher codon adaptation index (CAI), 25.4 kcal/mol more favorable minimum free energy (MFE), and 3.4% lower uridine content on average, while improving codon stabilization coefficient (CSC) in over 90% of benchmark proteins under matched constraints. These gains translate into designs that are predicted to be more efficiently translated, more structurally stable, and less immunogenic, while supporting continuous objective reweighting at inference time.
|
|
Scooped by
mhryu@live.com
February 13, 5:20 PM
|
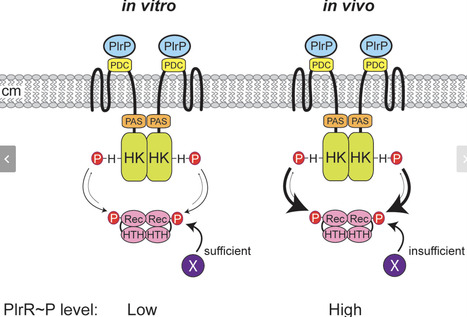
PlrSR, a member of the NtrYX family of two-component regulatory systems (TCSs), is required for the classical bordetellae, including the causative agent of whooping cough, Bordetella pertussis, to persist in the lower respiratory tract. The plrSR genes are in the middle of a six-gene cluster whose regulation and roles during infection were unknown. rsmB and plrP are often found 5′ to plrSR homologs in β- and γ-proteobacteria, while trkAH is often found 3′ to plrSR homologs in ⍺-proteobacteria. We investigated these genes to determine if they have a functional link to plrSR. We found that this gene cluster does not function as an operon. Rather, it contains two internal promoters: a weaker promoter in the 3′ end of rsmB and a stronger promoter in the 3′ end of plrS. Additionally, our results indicate that PlrP functions as a third component of the PlrSR TCS. Genetic manipulations of plrP, plrS, and plrR indicate that PlrP is essential in vitro and strongly suggest that it inhibits PlrS phosphatase activity, likely through PlrS’s PhoQ-DcuS-CitA (PDC) domain. Since our results indicate that PlrR can be phosphorylated by another unknown phosphodonor in vitro, limiting PlrS phosphatase activity ensures PlrR~P is not dephosphorylated to lethally low levels. Using natural-host models, we determined that high levels of PlrR~P are required for bacterial survival in the lower respiratory tract, and that PlrP affects PlrS activity in vivo. Given that plrP homologs always colocalize with ntrYX homologs, we propose that PlrP may fulfill similar functions in other β- and γ-proteobacteria that encode NtrYX-family TCSs, including nonpathogens.
|
|
|
Scooped by
mhryu@live.com
Today, 3:52 PM
|
Flavin mononucleotide (FMN) and flavin adenine dinucleotide (FAD) are essential redox cofactors in cellular metabolism. In this study, riboflavin-producing E. coli was engineered for de novo FMN and FAD synthesis. The RibFM-1 mutant with defective FAD synthase activity was overexpressed to produce FMN. NADH supply was enhanced by overexpressing sthA and deleting pntAB, while deletion of ndh encoding NADH dehydrogenase II and appB encoding cytochrome oxidase bd-II improved ATP regeneration. The resulting strain FMN08 produced 3457.3 mg/L FMN in fed-batch fermentation. FMNAT from Candida glabrata was engineered by semirational design to increase catalytic activity 2.14-fold, and coexpression of FMN1 from Saccharomyces cerevisiae enabled FMN-to-FAD conversion. Deletion of ushA encoding 5′-nucleotidase and overexpression of purA further promoted FAD production. The final strain FAD13 produced 2305.2 mg/L FAD in fed-batch fermentation. These results represent the highest reported titers of de novo synthesis of these redox cofactors.
|
|
Scooped by
mhryu@live.com
Today, 3:43 PM
|
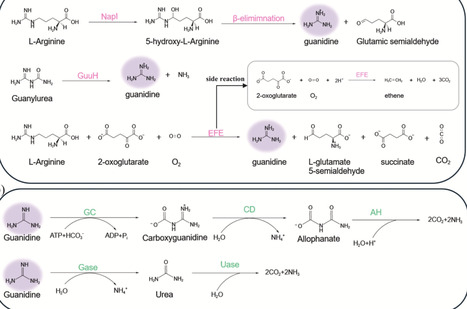
Guanidine is a chemically stable, nitrogen-rich moiety that constitutes a core structural element of numerous natural small molecules and is widely used in pharmaceuticals, animal feed, cosmetics, and related industries. In recent years, microbial cell factories have emerged as sustainable and efficient platforms for the production of guanidine derivatives. This review summarizes the central metabolic pathways of guanidine in microorganisms and provides critical insights for the metabolic engineering of guanidine derivatives. We highlight current biosynthetic strategies and recent advances in the production of representative guanidine derivatives, including L-arginine and its downstream products guanidinoacetate and agmatine. Finally, we discuss the key challenges limiting the efficient biosynthesis of guanidine and its derivatives and outline future directions toward industrially viable guanidine-based biomanufacturing.
|
|
Scooped by
mhryu@live.com
Today, 3:33 PM
|
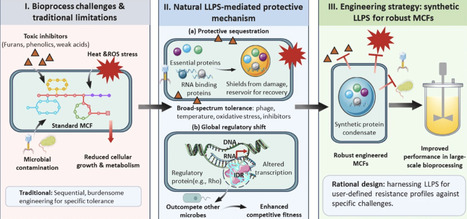
Microbial cell factories are pivotal platforms for converting lignocellulosic biomass into fuels and chemicals within the bioeconomy framework. Current bioproduction remains hindered by microbial sensitivity to inhibitors and process stresses, and by metabolic bottlenecks from poorly organized pathways and toxic intermediates that reduce titer, yield, and productivity. It is further limited by a narrow range of accessible bio-based chemicals. This review spotlights protein liquid–liquid phase separation (LLPS) as a powerful strategy to overcome these barriers. LLPS, a ubiquitous biophysical phenomenon, enables the formation of dynamic, membraneless condensates crucial for cellular functions like stress responses and metabolic channeling. Microbes leverage LLPS for protection against diverse stressors and to enhance metabolic efficiency by localizing enzymes and substrates. We critically assess how engineering microbial phase separation improves host fitness, streamlines metabolic flux to enhance economic performance, and enables in vivo assembly and functional integration of artificial metalloenzymes.
|
|
Scooped by
mhryu@live.com
Today, 3:25 PM
|
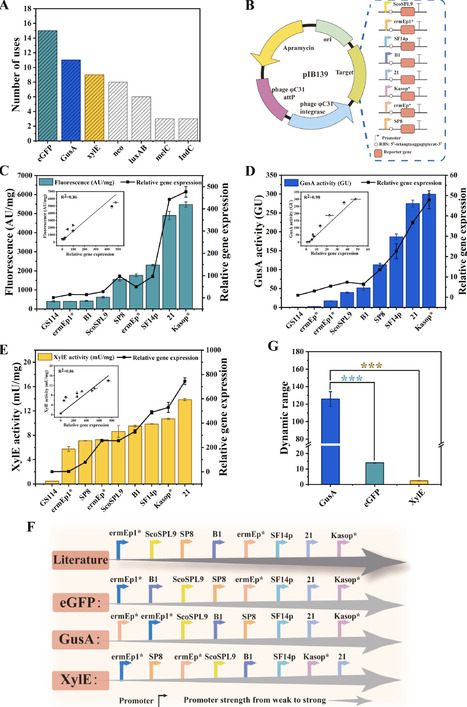
Streptomycetes are key producers of natural products (NPs), and engineering these organisms represents an effective strategy for enhancing NPs production. However, genetic elements (e.g., promoters and ribosome-binding sites (RBSs)) developed in wild-type or well-characterized model strains often exhibit limited adaptability in industrial strains, hindering their broader application. In this study, an efficient, low-cost strategy for constructing a gene expression element library (GEEL) for industrial Streptomyces was developed and validated using the ε-poly-ʟ-lysine (ε-PL) producing strain S. albulus GS114. Firstly, commonly used reporter genes were screened, and β-glucuronidase (gusA) was selected as the optimal reporter. Subsequently, a mixed ligation system (MLS) based on Gibson assembly was established to enable efficient assembly of pooled fragments into a vector. Using this system, libraries comprising 60 promoters and 30 RBSs were constructed. Each library was then introduced separately into S. albulus GS114 by single-round conjugation, and the resulting conjugants were screened for GUS activity. From these screens, 30 promoters and 14 RBSs spanning a range of strengths were identified, yielding GEELs specifically tailored to S. albulus GS114. Thirdly, five promoters and three RBSs were selected to optimize the expression of the pls gene, resulting in an ε-PL production of 4.73 g/L, reflecting a 2.13-fold increase compared to the control. Finally, this strategy was similarly applied to S. gilvosporeus to establish GEELs and optimize the expression of sgnM, leading to enhanced natamycin production. The customized GEEL construction strategy developed in this study provides a foundational toolkit for the precise regulation of key targets in industrial Streptomyces and the further enhancement of NPs biosynthesis.
|
|
Scooped by
mhryu@live.com
Today, 2:39 PM
|
It has long been debated in ecology whether communities behave as cohesive units or as loose collections of independent species. Here, we study this question in the context of community coalescence, the mixing of previously isolated communities, using bacterial microcosm experiments combined with ecological modeling. Our results demonstrate that interspecies interaction strength determines whether communities or species are the units of selection during coalescence. When interactions are moderate to strong, one parental community consistently outcompetes the other, indicating community-level selection. In contrast, under weak interactions, species fates are uncorrelated and the two communities contribute equally to the coalesced outcome, indicating the absence of community-level selection. These patterns extend to communities derived from natural samples with greater taxonomic diversity and richness. Furthermore, we identify two distinct regimes underlying community-level selection in experiments with different media conditions: an emergent regime in which collective dynamics shape outcomes that cannot be predicted from species traits alone, and a top-down regime where dominant species determine the winning community. Together, these results reconcile conflicting observations on community-level selection during community coalescence by demonstrating that communities behave as cohesive units only when interactions are sufficiently strong.
|
|
Scooped by
mhryu@live.com
Today, 2:08 PM
|
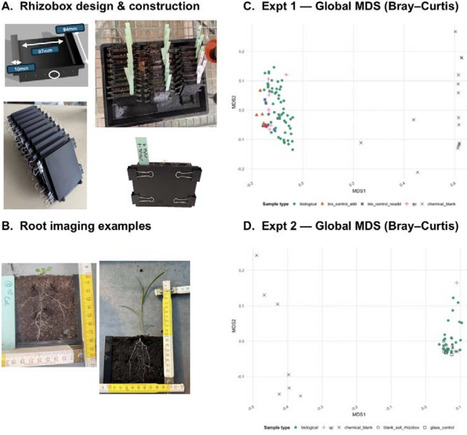
Root exudates play a central role in nutrient cycling, microbial recruitment, and plant-plant interactions, yet most experimental approaches for analyzing exudate chemistry rely on sterile hydroponic systems that poorly represent soil conditions. We present a low-cost, open-source, 3D-printed rhizobox platform and associated workflow that enable non-destructive root imaging and targeted rhizosphere soil sampling for LC-MS based metabolomics under realistic soil conditions. The design integrates a transparent removable window for repeated root observations, a defined soil volume to support spatially explicit sampling, and a blank-informed data-processing pipeline to distinguish plant-derived metabolites from soil and construction material background. We validated the system using the model plants Arabidopsis thaliana (Col-0) and Phragmites australis. We demonstrate reliable plant growth and consistent root development across the imaging window. We also show robust detection of species-specific rhizosphere metabolite profiles, with minimal variation in the vertical or temporal dimensions relative to the strong species effects. We further illustrate the application of the workflow in a factorial experiment manipulating social context (solo vs. conspecific pairs) and short-term heat stress in A. thaliana, showing that the approach is sensitive to treatment-associated changes in metabolite richness, diversity, and chemical composition in soil. The complete protocol, from rhizobox fabrication and assembly to soil extraction, LC-MS acquisition, and data curation can be implemented within 4-6 weeks using standard laboratory equipment and openly available design files. By combining ecological realism with analytical control, this workflow provides a broadly applicable method for quantifying rhizosphere metabolite dynamics across species, treatments, and spatial sampling zones, facilitating experimental studies of below-ground chemical processes in plant ecology.
|
|
Scooped by
mhryu@live.com
Today, 1:46 PM
|
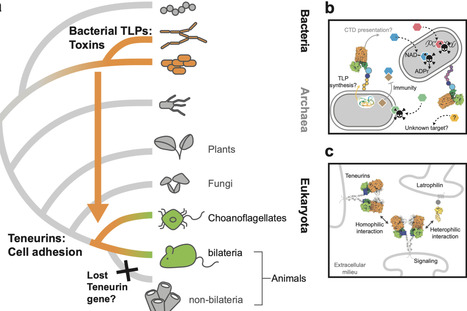
Horizontal gene transfer events were crucial in the emergence of multicellular life. A striking example is the acquisition of Teneurins, putative surface-exposed toxins in bacteria that function as cell adhesion receptors in metazoan neuronal development. Here, we demonstrate the evolutionary relationships between metazoan and bacterial Teneurins. We use cryogenic electron microscopy and bioinformatic analysis to show that bacterial Teneurins harbor a toxic protein in a proteinaceous shell. They are rare but widely distributed across bacterial taxa and are predominantly seen in species with complex social behaviors, suggesting roles in cell-to-cell interaction. This work confirms that metazoan Teneurins are repurposed bacterial toxins that have evolved to be essential mediators of intercellular communication in all advanced nervous systems. Their acquisition was a key event in the evolution of metazoans. Previous studies of Teneurins identified an uncharacterized family of Teneurin-like proteins in bacteria. Here, the authors show these proteins are widespread across both Gram groups but limited to certain species, where they form barrel-like structures that encapsulate a toxin and are co-expressed with potent immunity genes.
|
|
Scooped by
mhryu@live.com
Today, 1:19 PM
|
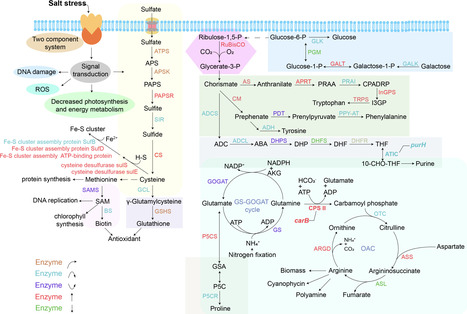
Cyanobacteria dominate the foundational biological soil crusts (biocrusts) of hypersaline deserts, but how they adapt to extreme hypersaline environments remains a fundamental question. Here, we assemble the genome and investigate the adaptation of the halotolerant cyanobacterium Nostoc sp. FACHB-892, a prevalent species of the Tengger Desert. Comparative phylogenomics across 76 cyanobacterial strains from diverse habitats reveals that this desert lineage underwent a significant expansion of genes involved in photosynthesis, signal transduction, and energy metabolism. Integrated transcriptomic and metabolomic analyses under salt stress demonstrate a concentration-dependent, multi-pathway response, critically involving the accumulation of key amino acid metabolism. We identify four genes putatively central to haloadaptation exhibiting signatures of adaptive convergence. Their essential roles in salt tolerance are confirmed through functional validation in E. coli. This study uncovers the coordinated genomic and metabolic mechanisms underpinning cyanobacterial resilience in one of Earth’s harshest environments.
|
|
Scooped by
mhryu@live.com
Today, 1:01 PM
|
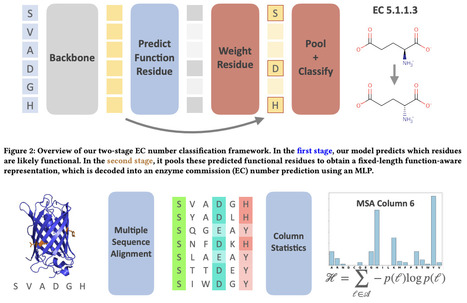
Predicting enzymatic function from a protein sequence is a fundamental task in protein discovery and engineering. In this paper, we present Semi-supervised Learning for Enzyme Classification (SLEEC): a semi-supervised learning framework that learns a function-aware protein representation for Enzyme Commision (EC) number prediction. SLEEC achieves SOTA performance on standard benchmarks and provides interpretable, residue-level annotations. We further demonstrate that our framework is robust to benign sequence modifications routinely observed in protein engineering workflows-such as appending functional tags- a desirable property that current ML frameworks lack. Our main technical contribution is a multiple sequence alignment (MSA)-based data augmentation technique for discovering sparse residue activations within a given enzyme sequence.
|
|
Scooped by
mhryu@live.com
Today, 12:22 PM
|
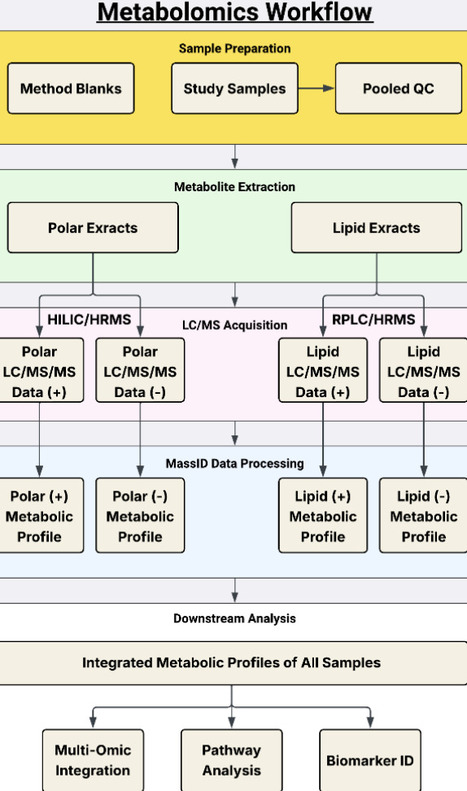
Liquid chromatography coupled to mass spectrometry (LC/MS) is a powerful tool in metabolomics research, generating tens-of-thousands of signals from a single biological sample. However, current software solutions for unbiased assessment of metabolomics data analysis are limited by complex sources of noise and non-quantitative metabolite identifications that make results difficult to interpret. Here, we present MassID, a cloud-based untargeted metabolomics pipeline that aims to overcome the innate challenges of unbiased metabolite analysis and perform end-to-end data processing, transforming raw spectra to normalized and identified metabolite profiles. MassID incorporates a suite of software functionalities, including deep learning-based peak detection and comprehensive noise filtering. In addition, with MassID we introduce a novel software module: DecoID2 that enables probabilistic metabolite identification for false discovery rate (FDR)-controlled metabolomics. When applied to a human plasma dataset, MassID results in near-complete signal annotation, identification of >4,000 metabolites (including >1,200 compounds at an FDR <5%) across four complementary LC/MS runs, and enables integrated downstream analyses to understand biochemical dysregulation at both the molecular and pathway level. When compared to the Metabolomics Standards Initiative (MSI) confidence levels, identification probability generally correlated with MSI levels. However, only 356/418 of MSI Level 1 compounds were identified with <5% FDR and the remaining 884 FDR < 5% compounds were identified from MSI L2-L3 compounds, highlighting the enhanced specificity and discovery potential achieved by MassID.
|
|
Scooped by
mhryu@live.com
Today, 12:16 PM
|
RNA structure plays a central role in how transcripts function, but inferring it reliably remains difficult, especially when pseudoknots need to be part of the prediction. Chemical probing experiments provide additional signals, yet these signals do not directly identify base pairing partners. RNA proximity ligation provides direct evidence of base pairing, but balancing this evidence with pseudoknot prediction accuracy and scalability of structure prediction for long sequences remains challenging. We present CPLfold, a fast and flexible RNA folding method that combines thermodynamic modeling with chimeric evidence from RNA cross-linking and ligation experiments, while naturally supporting pseudoknots. CPLfold scales to long sequences and recovers more accurate global structures and long-range interactions than existing approaches across multiple benchmarks such as COMRADES and IRIS. By tuning two simple trade-off parameters (α, β) the method allows flexibility in the level of incorporating chimeric evidence and asserting pseudoknots. Availability and Implementation: Source code and scripts are available at https://github.com/Vicky-0256/CPLfold.
|
|
Scooped by
mhryu@live.com
February 13, 5:24 PM
|
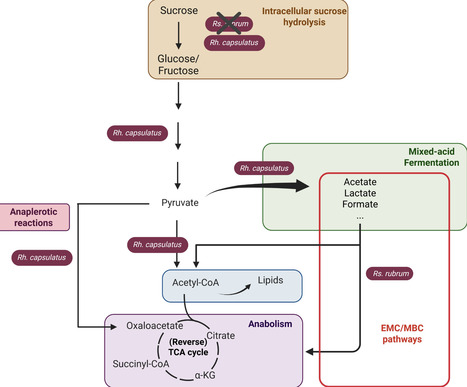
Purple non-sulfur bacteria (PNSB) are well known to have an exceptional metabolic versatility. However, while the growth of PNSB on sugar-rich streams has been extensively explored, their ability to metabolize sugars is poorly understood. Here, we explore the metabolic mechanisms of sucrose, glucose, and fructose utilization in two phototrophic PNSB, Rhodospirillum rubrum and Rhodobacter capsulatus. Our findings demonstrate distinct carbohydrate assimilation capacities, as well as the use of different metabolic strategies for each species. Moreover, a trophic link was identified between the two species during co-cultivation, resulting from the production of fermentation by-products by R. capsulatus, which are then reassimilated by R. rubrum. Finally, we demonstrate that the synergy observed between R. rubrum and R. capsulatus can be successfully scaled up in a photobioreactor system. Our study highlights how fundamental knowledge of metabolism and the establishment of a trophic link between two PNSB species might be useful for the development of biobased economy and resource recovery strategies.
|


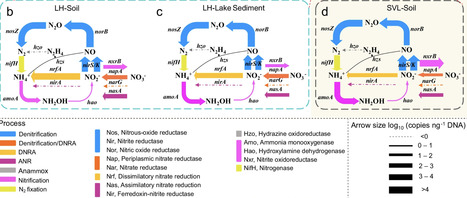











nitrogen pathway cycle, functional N-cycling genes encompass N2 fixation (nifH), nitrification (amoA, hao, and nxrB), denitrification (napA, narG, nirS/K, norB, and nosZ); dissimilatory nitrate reduction to ammonium (DNRA; nrfA), assimilatory nitrite reduction (ANR; nasA and nirA), and anaerobic ammonium oxidation (anammox; hzo)