Your new post is loading...

|
Scooped by
mhryu@live.com
February 5, 11:45 PM
|
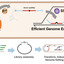
We used an autonomous lab, comprising a large language model (LLM) and a fully automated cloud laboratory, to optimize the cost efficiency of cell-free protein synthesis (CFPS). By conducting iterative optimization, the LLM-driven autonomous lab was able to achieve a 40% reduction in the specific cost ($/g protein) of CFPS relative to the state of the art (SOTA). This cost reduction was accompanied by a 27% increase in protein production titer (g/L). Iterative experimental design, experiment execution, data capture and analysis, data interpretation, and new hypothesis generation were all handled by the LLM-driven autonomous lab. The interface between OpenAI's GPT-5 LLM and Ginkgo Bioworks' cloud laboratory incorporated built-in validation checks via a Pydantic schema to ensure that AI-designed experiments were properly specified. Experimental designs were translated into programmatic specification of multi-instrument biological workflows by Ginkgo's Catalyst software and executed on Ginkgo's Reconfigurable Automation Cart (RAC) laboratory automation platform, with human intervention largely limited to reagent and consumables preparation, loading and unloading. By integrating LLMs with programmatic control of a cloud lab, we demonstrate that an LLM-driven autonomous lab can successfully perform a real-world scientific task, highlighting the potential of AI-driven autonomous labs for scientific advancement.
|
|
Scooped by
mhryu@live.com
February 5, 11:24 PM
|
Biomass separation represents a critical bottleneck in Komagataella phaffii-based biopharmaceutical processes, as typically high cell densities of 40 - 50 % create significant operational, technical and economic challenges for harvest operations. Yeast cell aggregation (flocculation) provides a solution to accelerate cell sedimentation by increasing particle size, thus allowing to improve biomass-supernatant separation efficiency during both natural gravity settling and (continuous) centrifugation operations. This study demonstrates successful engineering of K. phaffii strains with an inducible flocculation phenotype using CRISPR/Cas9-based genome editing to integrate the Saccharomyces cerevisiae FLO1 (FLO1) gene under control of various regulatory elements, including methanol-inducible and derepressible promoters. Flocculation strength could be enhanced by implementing transcriptional positive feedback circuits based on the methanol-inducible AOX1 promoter. To address methanol-free production requirements, we developed alternative systems to retrofit PAOX1-based ScFLO1 expression and exploited the derepressible PDF promoter, offering broader compatibility with biopharmaceutical manufacturing facilities. Flocculating cells cultivated in a bioreactor demonstrated significantly improved sedimentation behavior, with considerably lower supernatant turbidity after short low-speed centrifugation compared to non-flocculating controls. Crucially, cell flocculation had no negative impact on product amount and quality when expressing a multivalent NANOBODY® VHH molecule with pharmaceutical relevance. Thus, this work establishes the first genetically engineered flocculation system in K. phaffii compatible with recombinant protein production, providing the basis for an innovative approach to streamline harvest operations in biopharmaceutical processes.
|
|
Scooped by
mhryu@live.com
February 5, 11:14 PM
|
In synthetic biology, DNA assembly is a routine process where increasing demands for standardization, high-throughput capacity, and error-free execution are driving the development of accessible, automated solutions. Here, we present Slowpoke, a user-friendly and flexible workflow for Golden Gate-based cloning designed for the popular entry-cost, open-source liquid-handling platforms Opentrons OT-2 and Flex. Slowpoke automates the key steps of the DNA assembly process, including cloning, E. coli transformation, plating, and colony PCR, requiring user intervention primarily for colony picking and plate transfers. To further simplify the usage, we developed a free graphical user interface (GUI), available at https://slowpoke.streamlit.app/, which enables rapid protocol generation through simple file uploads. We validated the workflow using two Golden Gate-based toolkits, the MoClo Yeast Toolkit (YTK), and SubtiToolKit (STK). High assembly efficiencies were achieved across platforms for basic transcript unit constructions: 17/17 positive colonies with YTK on OT-2, 11/12 on Flex, and 8/13 with STK on OT-2. High-throughput assemblies were also performed with six parts in Flex using YTK-compatible parts, and 55 out of 57 combinations resulted in correct constructs. These results confirm the robustness and adaptability of the workflow across toolkit complexity and automation platforms. The Slowpoke suite, including code scripts and templates, is freely available at https://github.com/Tom-Ellis-Lab/Slowpoke, offering an accessible and modular solution for automating Golden Gate cloning in synthetic biology laboratories.
|
|
Scooped by
mhryu@live.com
February 5, 11:06 PM
|
The expansion of sequencing technologies and bioinformatics has greatly advanced our understanding of microbial “dark matter,” yet the isolation of pure cultures, especially among Archaea, remains rare and challenging. Cultivation is still essential for the reliable characterization of microbial metabolism, which cannot be fully replaced by metagenomics and other omics-based approaches. Here, we report the first cultivated representatives of a deep-branching archaeal lineage previously known as Candidatus Marsarchaeota. Our phylogenomic analyses place these isolates within the phylum Thermoproteota as a novel order, Tardisphaerales. Members of Tardisphaerales dominate the prokaryotic communities in acidic hot springs below 70°C, comprising up to 40% of the total microbial population, underscoring their ecological significance. Functional genomics and culture experiments reveal a thermoacidophilic, anaerobic lifestyle, with energy metabolism based on carbohydrate fermentation, particularly of polysaccharides. This metabolic capability is supported by numerous glycosidase-encoding genes and by unprecedented metabolic versatility among thermoacidophiles. The isolates possess complete glycolysis, Entner-Doudoroff, and pentose-phosphate pathways, allowing them to utilize different sugars. Specialization in polysaccharide hydrolysis presumably provides an adaptive advantage for these slow-growing archaea, as most other heterotrophic thermoacidophiles prefer peptides or simple sugars. Furthermore, robust defense mechanisms against reactive oxygen species and persistence in acidic conditions enable Tardisphaerales to outcompete other heterotrophs and maintain dominance in these extreme habitats. The discovery and cultivation of this new order expand prokaryotic taxonomy and reveal the key players in carbon cycling in acidic geothermal ecosystems.
|
|
Scooped by
mhryu@live.com
February 5, 8:56 PM
|
Photosymbioses provide carbon and oxygen to the biosphere, yet the mechanisms underlying their evolution remain poorly understood. We develop a naive system based on the predatory ciliate Tetrahymena thermophila, not known for hosting symbionts, to recapitulate early events of photosymbiosis evolution. T. thermophila readily phagocytoses eukaryotic algae (Chlorella variabilis) or cyanobacteria (Synechococcus elongatus). Feeding on either prey in a low-carbon medium provided little or no growth advantage. By contrast, in a hypoxic environment, both intracellular C. variabilis and S. elongatus can support temporary survival of T. thermophila. These results suggest that oxygen supply within the host could represent a more plausible initial advantage supporting photosymbiosis evolution than carbon metabolites. While most extant photosymbioses are based on carbon supply to the host cell, we therefore propose that this would be a secondary event occurring from initial evolution in anoxic or hypoxic conditions, where O2 production is crucial for establishing the initial steps of photosymbiosis.
|
|
Scooped by
mhryu@live.com
February 5, 8:40 PM
|
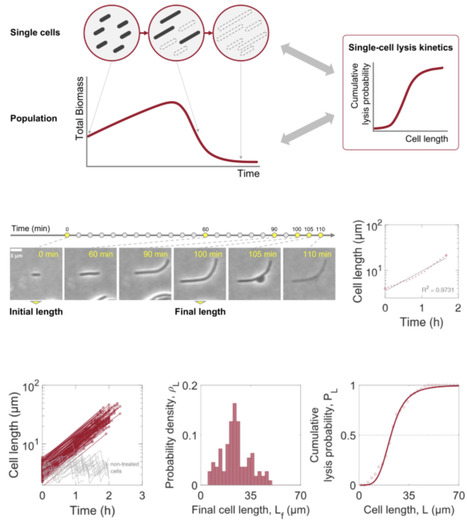
Treatment of sensitive bacteria with beta‐lactam antibiotics often leads to two salient population‐level features: a transient increase in total population biomass before a subsequent decline, and a linear correlation between growth and killing rates. However, it remains unclear how these population‐level responses emerge from collective single‐cell responses. During beta‐lactam treatment, it is well‐recognized that individual cells often exhibit varying degrees of filamentation before lysis. We show that the cumulative probability of cell lysis increases sigmoidally with the extent of filamentation and that this dependence is characterized by unique parameters that are specific to bacterial strain, antibiotic dose, and growth condition. Modeling demonstrates how the single‐cell lysis probabilities can give rise to population‐level biomass dynamics, which were experimentally validated. This mapping provides insights into how the population biomass time‐kill curve emerges from single cells and allows the representation of both single‐ and population‐level responses with universal parameters.
|
|
Scooped by
mhryu@live.com
February 5, 8:12 PM
|
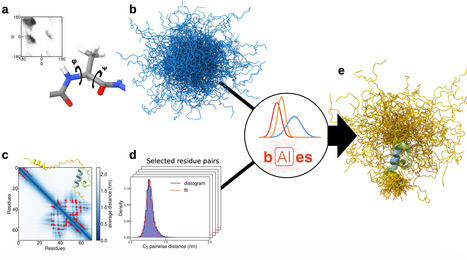
Intrinsically disordered proteins are ubiquitous in biological systems and play essential roles in a wide range of biological processes and diseases. Despite recent advances in high-resolution structural biology techniques and breakthroughs in deep learning-based protein structure prediction, accurately determining structural ensembles of IDPs at atomic resolution remains a major challenge. Here, we introduce bAIes, a Bayesian framework that integrates AlphaFold2 predictions with physico-chemical molecular mechanics force fields to generate accurate atomic-resolution ensembles of IDPs. We show that bAIes produces structural ensembles that match a wide range of high- and low-resolution experimental data across diverse systems, achieving accuracy comparable to atomistic molecular dynamics simulations but at a fraction of their computational cost. Furthermore, bAIes outperforms state-of-the-art IDP models based on coarse-grained potentials as well as deep-learning approaches. Our findings pave the way for integrating structural information from modern deep-learning approaches with molecular simulations, advancing ensemble-based understanding of disordered proteins. Here, the authors present bAIes, which integrates AlphaFold2 information into a molecular dynamics framework to efficiently generate atomic-resolution ensembles of intrinsically disordered proteins.
|
|
Scooped by
mhryu@live.com
February 5, 8:05 PM
|
Plant growth-promoting rhizobacteria (PGPR) are widely recognized as sustainable agricultural inputs because they enhance nutrient availability, reduce reliance on chemical fertilizers, and improve soil and plant health. However, their field performance is often limited by reduced viability and stability under environmental stresses. This study aimed to develop and compare double-layer and multilayer polysaccharide–protein–based encapsulation systems to improve the protection, long-term stability, and controlled release of PGPR. Pseudomonas fluorescens T17-4 and Bacillus velezensis VRU1 were encapsulated using an alginate–whey protein isolate core, with successive outer layers of apricot gum and pectin to form double-layer and multilayer capsules. Mesoporous silica nanoparticles were added to further reinforce the capsule structure. The multilayer encapsulation system outperformed the double-layer formulation, showing higher encapsulation efficiency (> 90%), improved structural integrity, enhanced long-term stability with over 90% bacterial viability after six months, and a more controlled release profile. These findings demonstrate that multilayer polysaccharide–protein encapsulation is an effective strategy for protecting PGPR and ensuring their sustained delivery, highlighting its potential for developing robust biofertilizers in sustainable agriculture.
|
|
Scooped by
mhryu@live.com
February 5, 7:57 PM
|
Plants encounter a myriad of microorganisms that can invade with detrimental or beneficial outcomes. Taking a membrane-centric point of view, we discuss the cellular events underlying microbe-induced cell signaling, the navigation of microbes within plant tissues, and the molecular exchanges occurring at plant–microbe interfaces. We discuss the implications of individual membrane lipids and the emerging role of membrane biophysics in non-self and modified-self sensing. We highlight the common themes underlying the active role of membranes during plant interactions with viruses, bacteria, oomycetes, and fungi, and define exciting directions for future research.
|
|
Scooped by
mhryu@live.com
February 5, 7:44 PM
|
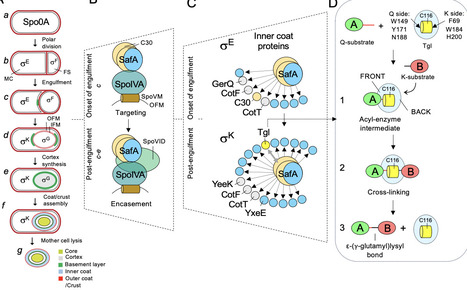
Hub proteins are highly connected nodes in protein-protein interaction networks and are often intrinsically disordered proteins (IDPs) or contain intrinsically disordered regions. In Bacillus subtilis, the morphogenesis of the spore surface is orchestrated by a set of so-called morphogenetic proteins that guide the assembly of distinct layers. Formation of the inner coat is directed by SafAFL and its shorter isoform, C30. Both are expressed early in sporulation under the control of σE and localize at the interface between the developing inner coat and the underlying cortex peptidoglycan. From this site, they act as organizational hubs, recruiting client proteins essential for coat maturation. Among these is Tgl, a transglutaminase synthesized later in development following activation of σK after engulfment completion. We show that the C30 domain exhibits IDP-like features yet self-assembles into >1200 kDa complexes stabilized by disulfide bonds and that these bonds are required for subsequent proper Tgl-mediated “spotwelding” cross-linking. Small-angle X-ray scattering (SAXS) and photobleaching show that Tgl immobilizes but does not drastically alter these assemblies. These findings support a hierarchical, biphasic model for inner coat assembly: initial self-assembly and disulfide stabilization, followed by Tgl-mediated cross-linking and structural stabilization. According to this model, the forms of SafAFL/C30 that dominate the two stages recruit different client proteins in register with the course of morphogenesis.
|
|
Scooped by
mhryu@live.com
February 5, 2:52 PM
|
ssDNA viruses are important components of diverse ecosystems; however, it remains challenging to systematically identify and classify them. This is partly due to their broad host range and resulting genomic diversity, structure and rapid evolutionary rates. In addition, distinguishing genuine ssDNA genomes from contaminating sequences in metagenomic datasets (e.g. from commercial kits) has been an unresolved issue for years. Here, we present CRESSENT (CRESS-DNA Extended aNnotation Toolkit), a comprehensive and modular bioinformatic pipeline focused on ssDNA virus ‘genome-to-analysis’ and annotation. The pipeline integrates multiple functionalities organized into several modules: sequence dereplication, decontamination, phylogenetic analysis, motif discovery, stem-loop structure prediction and recombination detection. Each module can be used independently or in combination with others, allowing researchers to customize their analysis workflow. With this tool, researchers can comprehensively and systematically include ssDNA viruses in their viromics workflows and facilitate comparative genomic studies, which are often limited to dsDNA viruses, therefore leaving behind a crucial component of the microbiome community under study. Benchmarking analyses demonstrated that CRESSENT efficiently processes ssDNA virus datasets of varying scales, completing small family-level analyses within minutes and moderate comparative genomics studies within hours using standard computing resources. Its modular, parallelized design ensures scalability and low memory usage, making it accessible to research groups with diverse computational capacities.
|
|
Scooped by
mhryu@live.com
February 5, 1:05 PM
|
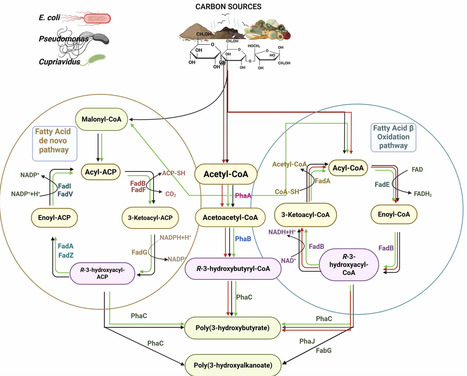
Polyhydroxyalkanoates (PHAs) are biodegradable polymers produced by various microorganisms as intracellular carbon and energy reserves. Their potential to replace petroleum-based plastics has made them central to sustainable materials research. However, their large-scale commercialization is hindered by high production costs, prompting efforts to improve yield and identify low-cost, non-food carbon sources. This review examines PHA biosynthesis optimization via advances in microbial strain-substrate selection ensuring economic feasibility. Focus is placed on three key bacterial genera, Cupriavidus, Pseudomonas, and E. coli analyzing their metabolic flexibility, suitable substrate range and PHA content (g product/ g substrate). E. coli species and C. necator demonstrate high polymer contents from largely simple sugars and fatty-acid substrates respectively, while Pseudomonas species offer a broad substrate adaptability, particularly waste streams. The environmental impact assessment of some C. necator and Pseudomonas strains highlight low carbon footprint from waste lipids compared to PHA production from glucose or bottle-grade PET, however these results are contingent to specific scenarios without a reproducible and quantifiable GWP values across these genera. Integrating microbial engineering of agro-industrial and organic wastes in adherence to regulatory frameworks around waste valorization and optimization, genus-specific LCA bioprocessing approach offers a path toward economically viable and environmentally sustainable PHA production.
|
|
Scooped by
mhryu@live.com
February 5, 12:57 PM
|
Archaea, a domain of microorganisms found in diverse environments, including the human microbiome, represent the closest known prokaryotic relatives of eukaryotes. This phylogenetic proximity positions them as a relevant model for investigating the evolutionary origins of nucleic acid secondary structures such as G-quadruplexes (G4s) which play regulatory roles in transcription and replication. Although G4s have been extensively studied in eukaryotes, their presence and function in archaea remain poorly characterized. In this study, a genome-wide analysis of the halophilic archaeon Haloferax volcanii identified over 5800 potential G4-forming sequences. Biophysical validation confirmed that many of these sequences adopt stable G4 conformations in vitro. Using G4-specific detection tools and super-resolution microscopy, G4 structures were visualized in vivo in both DNA and RNA across multiple growth phases. Comparable findings were observed in the thermophilic archaeon Thermococcus barophilus. Functional analysis using helicase-deficient H. volcanii strains further identified candidate enzymes involved in G4 resolution. These results establish H. volcanii as a tractable archaeal model for G4 biology.
|
|
|
Scooped by
mhryu@live.com
February 5, 11:35 PM
|
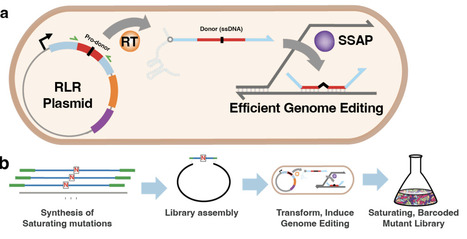
Saturation mutagenesis is a powerful tool for understanding and engineering the function of biological systems, and has been applied successfully to characterize the mutational landscape of individual proteins and genetic loci. However, it has not been applied at the whole-genome scale due to the challenges of both creating and quantifying a saturating set of mutations. Here we introduce Biobloom, a retron-based method for barcoded saturation mutagenesis at the scale of a whole bacterial genome. We constructed a barcoded Biobloom library with >99% projected sampling of saturating single-nucleotide polymorphism (SNP) mutations of the E. coli genome, and applied it to identify beneficial mutations under salt and antibiotic selection. Relative to other techniques like CRISPR-enabled mutagenesis or Adaptive Laboratory Evolution, Biobloom excels at identifying diverse causal SNPs quickly and at smaller working volumes. A barcoded, saturating mutation library is also a shared resource, and we are releasing the updated Biobloom-E.coli-2.0 library to the scientific community for broader adoption and application. Together, Biobloom makes barcoded saturation mutagenesis accessible at whole-genome scale, creating new opportunities for large-scale data collection and bacterial engineering.
|
|
Scooped by
mhryu@live.com
February 5, 11:20 PM
|
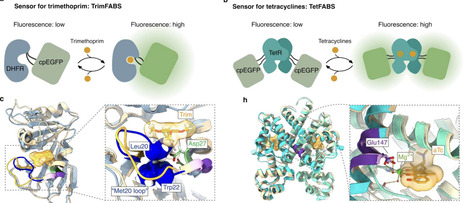
Antibiotic treatment can fail due to insufficient drug availability at the site of infection or limited accumulation within bacterial pathogens. However, it is poorly understood how antibiotics penetrate infected tissues and complex bacterial aggregates, limiting insights into the mechanisms of treatment failure. Here, we present genetically-encoded allosteric biosensors for two antibiotic classes, trimethoprim and tetracycline, which enable real-time monitoring of antibiotic concentrations inside bacterial cells. The biosensors consist of circularly permuted EGFP linked to the sensory domains DHFR or TetR. To extend this approach to low oxygen environments, we engineered an oxygen-independent trimethoprim biosensor by fusing DHFR to a circularly permuted version of the fluorogenic protein FAST. Using these biosensors, we monitored the antibiotic exposure dynamics of intracellular Salmonella enterica during macrophage infection at the single-cell level, and antibiotic penetration into anaerobic regions of Vibrio cholerae biofilms, as well as antibiotic availability in microoxic conditions in a human bladder tissue model infected with uropathogenic Escherichia coli. These fluorescent biosensors have the potential to be broadly applied for determining antibiotic distributions at infection sites with high spatial and temporal resolution.
|
|
Scooped by
mhryu@live.com
February 5, 11:10 PM
|
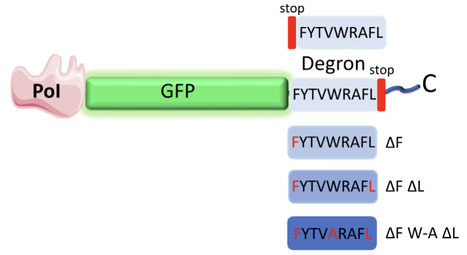
Targeted protein degradation relies on short sequence elements, termed degrons, that direct proteins to the cellular degradation machinery. Here, we report a nine-amino-acid degron (FYTVWRAFL) derived from the 3′ untranslated region (UTR) of human MTCH2 and demonstrate its utility as a tuneable protein-degradation tool. When appended to the C-terminus of proteins, FYTVWRAFL induces rapid and robust degradation by the ubiquitin-proteasome system. Using pharmacological inhibition and genetic perturbation, we show that degradation mediated by this degron is dependent on the E3 ubiquitin ligase MDM2. Structural modeling revealed that FYTVWRAFL interacts with the hydrophobic cleft of MDM2 in a manner similar to the p53 degron. Three hydrophobic residues form the core interaction interface. By systematic mutagenesis of these residues, we generated a panel of degron variants that confer graded levels of protein stability. We demonstrate the versatility of this system by achieving tunable expression of the endogenous protein Elm1 in Saccharomyces cerevisiae. Collectively, our study establishes a compact, transportable, and tunable degron system as a robust toolkit for quantitative control of protein abundance across eukaryotic systems.
|
|
Scooped by
mhryu@live.com
February 5, 9:00 PM
|
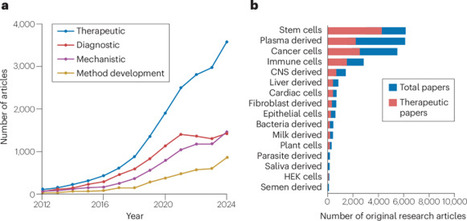
Owing to their natural origin and biocompatibility, extracellular vesicles (EVs) are being recognized as next-generation vehicles for targeted drug delivery. Despite their potential as therapeutic carriers, EVs suffer from heterogeneity, low yields, limited cargo loading efficiency and rapid clearance by the mononuclear phagocyte system. Since the first EV-based clinical trial in 2005, more than 100 clinical trials have investigated the use of EVs as therapeutics and drug carriers. Despite this, no EV-based therapies have received regulatory approval to date. This gap between preclinical research activity and clinical translation underscores persistent scientific challenges and regulatory hurdles that continue to impede the advancement of EV-based therapeutics. In this Review, we examine the research articles published in the field between 2012 and 2024 (38,177 articles), highlighting key developments, persistent challenges and evolving assumptions. We review the current EV landscape and clinical trials, focusing on their organotropism and use as carriers for therapeutics. We compare their advantages and limitations in relation to other nanoparticles, such as lipid nanoparticles and liposomes, and examine how labelling strategies and cell sources influence EV biodistribution. Finally, we outline translational considerations for EV-based therapeutics and propose additional reporting standards, complementing the MISEV 2023 guidelines. Extracellular vesicles (EVs) are emerging as biocompatible carriers for targeted drug delivery, yet challenges in yield, cargo loading and biodistribution persist. In this Review, key trends from over a decade of research are analysed, comparing EVs with lipid-based systems and outlining strategies for improving therapeutic translation and reporting standards.
|
|
Scooped by
mhryu@live.com
February 5, 8:49 PM
|
Flavonoids, produced by the plant under nutrient stress, are required to initiate the legume-rhizobia symbiosis through the activation of rhizobial nod genes. Notwithstanding the central role of flavonoids in nodulation, their transcriptional regulation remains poorly understood. Here, we show that the nodulation signaling pathway 2 (NSP2) is required for transcriptional activation of flavonoid biosynthesis genes during nodulation in Medicago truncatula. Furthermore, MYB40, a legume-specific MYB transcription factor, is induced by rhizobia in the root epidermis. MYB40 directly binds to flavonoid biosynthetic gene promoters and is required for normal levels of nodulation. Biochemical and genetic evidence reveal that NSP2, not NSP1, interacts with MYB40 during rhizobial infection to strongly upregulate the symbiotic gene chalcone O-methyltransferase 1 in a manner dependent on MYB40 binding sites. Moreover, the overexpression of MYB40 and a microRNA-resistant NSP2 variant enhances nodulation under suboptimal rhizobial availability, suggesting this module fine-tunes symbiosis efficiency. Additionally, flavonoid regulation by NSP2 and MYB40 appears to facilitate arbuscular mycorrhizal colonization under nutrient starvation. Together, our findings establish an NSP2-MYB40 module that integrates symbiotic signaling with metabolic reprogramming, representing an evolutionary innovation for optimizing nitrogen acquisition in dynamic environments.
|
|
Scooped by
mhryu@live.com
February 5, 8:31 PM
|
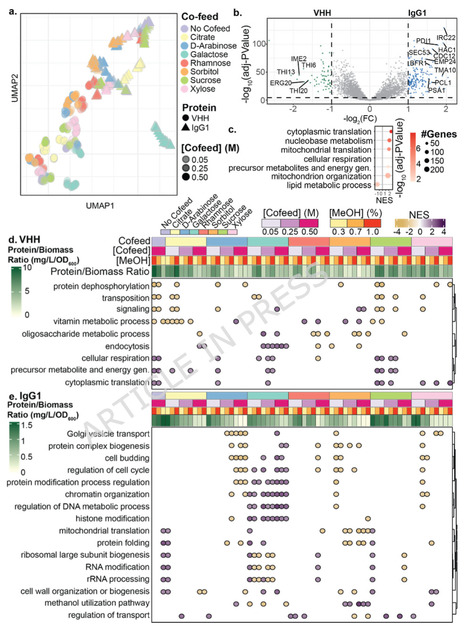
Demand for recombinant proteins is rapidly growing, driven by their use as biotherapeutics, vaccine components, industrial enzymes, and food ingredients. The growing market requires novel strategies for increasing protein production in cellular hosts. Systems-level frameworks have been used to improve production, but have had difficulty relating complex cellular pathways with protein expression. Here, we demonstrate a method for mapping relationships between gene expression signatures and carbon source-related phenotypes related to recombinant protein production. Our approach induces systematic perturbations in cultures of K. phaffii using varied co-feeds of carbon sources. The different carbon sources significantly impacted cell growth, specific productivity, and transcriptional states. With these data, we identified metagenes for both immunoglobulin G1 monoclonal antibody (IgG1) and Variable domain on a heavy chain (VHH) antibody that explained significant transcriptomic variance. These metagenes strongly associated with two phenotypes: production of recombinant protein-to-biomass ratio, and response to methanol induction. We used these results to identify and knockout 31 novel gene targets for which expression inversely correlated with productivity. Nine of these genes improved productivity of IgG1 by up to 3x and ten genes increased productivity of VHH by up to 1.7x. Many of these genes are involved in the modulation and progression of the cell cycle but interestingly, disruption had little to no impact on cell growth. This study establishes a framework for relating gene signatures to complex cellular phenotypes, providing a robust methodology for assessing production processes and identifying new targets for cellular engineering. While the identified specific metagenes depend on the complexity and structure of the recombinant protein produced, this framework is extensible across diverse proteins and potentially other host organisms. These signatures may serve as scale-independent, cellular-level metrics for traits like efficiency of production of recombinant proteins, facilitating the translation of findings across different scales and cultivation modes. Furthermore, this framework enables the identification of novel targets for genomic modifications that can improve strain performance, offering a predictive tool for the rational design of high-performing microbial cell factories.
|
|
Scooped by
mhryu@live.com
February 5, 8:09 PM
|
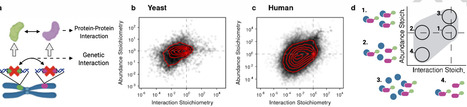
Genetic interaction and protein-protein interaction networks have proved among the most important tools for inference of gene function and genotype-phenotype relationship. It remains unclear, however, how these two networks are related to each other. Here we demonstrate that the strengths of epistatic genetic interactions between two genes strongly correlate with the binding free energies of interactions between their cognate proteins, for both yeast and human genomes. Consequently, we show that genetic interaction and protein-protein interaction networks can reciprocally predict each other. Further, we observe that functional divergence and redundancy in duplicated genes (paralogs) are reflected in the binding affinity of their interactions with other proteins and in their genetic interaction strengths. Finally, we demonstrate that the overall topologies of genetic interaction and protein-protein interaction networks significantly overlap in two topological features: modules, in which genes/proteins are organized by common function, and connectors, which link modules to each other. This opens avenues for integrated network approaches in understanding cellular processes and flow of genomic information. Genetic and protein-protein interaction networks can be used to infer gene function and genotype-phenotype relationships. Here, the authors show that genetic interaction patterns reflect protein binding affinities.
|
|
Scooped by
mhryu@live.com
February 5, 8:01 PM
|
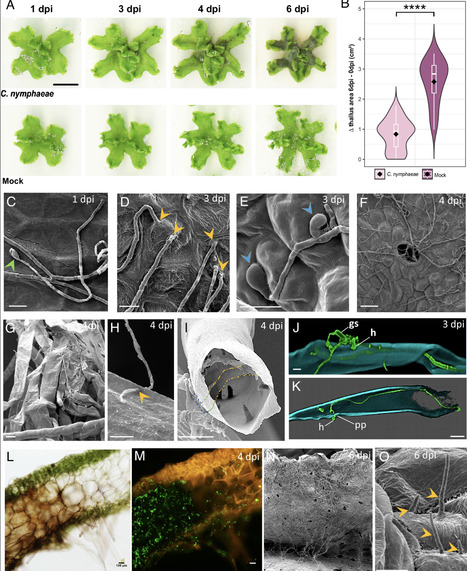
The liverwort Marchantia polymorpha has emerged as a model for studying plant immunity in bryophytes, providing unique insights into conserved defense mechanisms across land plants. By contrast, Marchantia-specific immune mechanisms remained largely underexplored. In this study, we investigated the genetic basis of quantitative resistance in M. polymorpha against the fungal pathogen Colletotrichum nymphaeae, a naturally occurring compatible parasite. Through a combination of phenotypic, cytological, and transcriptomic approaches, combined with genome-wide association studies (GWAS), we identified key defense-related genes and pathways. Leveraging the biological and genetic variability present in a collection of natural M. polymorpha accessions, we highlight the role of horizontally transferred microbial-like terpene synthase genes, which may contribute to the exceptional terpene diversity of liverworts and potentially play a role in pathogen resistance. GWAS uncovered candidate loci associated with resistance traits, implicating both core immune components and specialized metabolic pathways. Transcriptomic analyses performed on two accessions with contrasting phenotypes after inoculation with C. nymphaeae revealed the upregulation of accession-specific and horizontally acquired genes. These results provide insights into the specific molecular underpinnings of bryophyte immunity and underscore the evolutionary significance of horizontal gene transfer and specialized metabolites in shaping plant–pathogen interactions.
|
|
Scooped by
mhryu@live.com
February 5, 7:47 PM
|
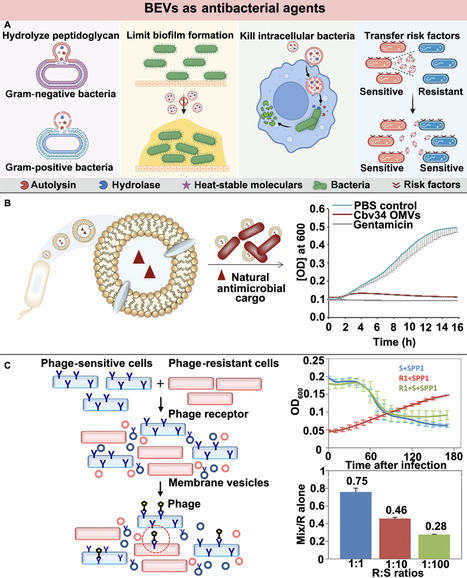
The overuse and misuse of antibiotics have led to widespread resistance in bacteria, which makes infections difficult to treat. The insufficient prevention measures, limited treatment options, and delayed antibiotic developments call for immediate global actions to discover effective and safe treatments for bacterial infections. Over the past decades, more and more studies have found that bacterial extracellular vesicles (BEVs) secreted by bacteria with nanoscale size, lipid bilayer structure, pathogen-associated molecular patterns, and inherent bioactive substances are the ideal candidates for bacterial infection treatment. Meanwhile, advanced engineering approaches have further endowed these BEVs with more customizable properties to effectively fight against bacterial infections. Herein, the present review begins with an overview of the biogenesis and biocomponents of BEVs to better comprehend their bioactivities against bacterial infections. Their isolation and engineering approaches are then introduced, with an emphasis on the diverse genetic, physical, and chemical strategies to functionalize them with desirable capacities for the optimal treatment of bacterial infections. Recent advances in exploring the natural BEVs as antibacterial and antiadhesion agents, as well as the engineered BEVs as vaccine antigens, vaccine adjuvants, and delivery nanocarriers, are expounded successively. Discussions on the new trend of engineering BEVs as nanoweapons to combat bacterial infections, in terms of advantages and challenges, are provided at the end to expedite these BEV-based therapeutic modalities for bacterial infections from bench to bedside.
|
|
Scooped by
mhryu@live.com
February 5, 6:54 PM
|
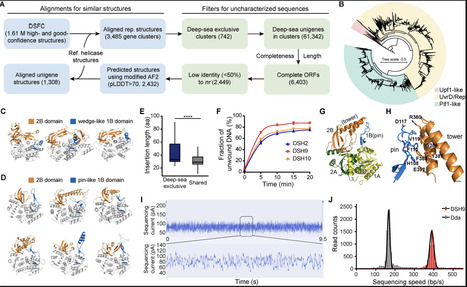
The deep sea as the largest and maybe most hostile environment on Earth is still underexplored especially regarding its genetic repertoire. Yet, previous work has revealed significant habitat-specific deep-sea biodiversity. Here, we present an integrated deep-sea genetic dataset comprising 502 million nonredundant genes from 2,138 samples and 2.4 million predicted structures, and used it to link specific protein structures with genetic variants associated with life in the deep sea and to assess their biotechnology potential. Combining global sequence analysis with biophysical and biochemical measurements revealed unprecedented sequence diversity, yet substantial structural conservation of proteins. Especially proteins involved in replication, recombination, and repair were identified to be under rapid evolution and with specialized properties. Among these, a structurally divergent helicase exhibited advantages in controlling nanopore sequencing speed. Thus, our work positions the deep sea as a unique evolutionary engine that generates and hosts genetic diversity and bridges genetic knowledge with biotechnology.
|
|
Scooped by
mhryu@live.com
February 5, 1:21 PM
|
Sparked by innovations in generative artificial intelligence (AI), the field of protein design has undergone a paradigm shift with an explosion of new models for optimizing existing enzymes or creating them from scratch. After more than one decade of low success rates for computationally designed enzymes, generative AI models are now frequently used for designing proficient enzymes. Here, we provide a comprehensive review and classification of generative AI models for enzyme design, highlighting models with experimental validation relevant to real-world settings and outlining their respective limitations. We argue that generative AI models now have the maturity to create and optimize enzymes for industrial applications. Wider adoption of generative AI models with experimental feedback loops can speed up the development of biocatalysts and serve as a community assessment to inform the next generation of models.
|
|
Scooped by
mhryu@live.com
February 5, 1:01 PM
|
Bacterial species differ dramatically in their growth rates, reflecting distinct ecological strategies and physiological constraints. Because protein synthesis is a major determinant of cellular replication, I examined how genomic investment in the translation machinery varies across bacteria with widely different doubling times. Using 20 bacterial species spanning two major bacterial kingdoms (Bacillati and Pseudomonadati), I quantified ribosomal RNA (rrn) operon number, total tRNA gene number, and the allocation of tRNA genes among amino acids. Rapidly replicating Vibrio natriegens has 11 rrn operons and 129 tRNA genes in its genome, whereas slowly replicating Borrelia burgdorferi has only one rrn operon and 32 tRNA genes. I show that both the rrn operon number and the tRNA gene number decline sharply with increasing generation time, a pattern observed independently within each bacterial kingdom. Moreover, tRNA gene allocation is highly non-uniform: amino acids that are frequently used in proteins and encoded by large synonymous codon families are supported by disproportionately more tRNA genes. This relationship is well described by a simple model incorporating amino acid usage and codon family size, as illustrated in rapidly growing species such as Vibrio natriegens and Clostridium perfringens. In contrast, slow-growing bacteria maintain relatively minimalist translation systems. Together, these results demonstrate that bacterial genomes are systematically optimized for translation in a manner tightly coupled to growth strategy, revealing how natural selection tunes both the capacity and structure of the translation machinery.
|