Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 1:10 AM
|
Despite growing evidence that viruses exploit liquid–liquid phase separation (LLPS), the significance of LLPS during infection remains elusive. Two recently published papers reveal that plant viruses use LLPS to reprogram host systems, promoting replication and immune suppression. These studies redefine LLPS as an active regulatory hub in plant–virus interactions.
|
|
Scooped by
mhryu@live.com
Today, 1:02 AM
|
Plant natural products (PNPs) offer exceptional chemical diversity and therapeutic potential, but their low natural abundance and complex biosynthetic origins often hinder scalable access. Microbial heterologous reconstruction has enabled the production of select PNPs, yet major challenges remain, including incomplete pathway elucidation, limited enzyme performance, and poor metabolic compatibility. Emerging advances in artificial biosynthesis provide a complementary strategy to transcend the constraints of native plant metabolism. By leveraging enzyme promiscuity, precursor prefunctionalization, modular pathway design, and recruitment of repurposed or engineered catalysts, artificial biosynthesis enables streamlined, controllable, and evolvable routes to structurally complex PNP scaffolds. These innovations define a rapidly advancing paradigm in which PNPs can be biosynthesized through predictive, design-driven, and non-natural pathways, offering new opportunities for discovery and sustainable biomanufacturing. bgc
|
|
Scooped by
mhryu@live.com
Today, 12:56 AM
|
Nucleotide sequences in the FASTQ or BAM format are widely shared, yet derived from platform-specific raw data outputs that differ across sequencing platforms. In Oxford Nanopore Technologies (ONT) sequencing, raw signal data contain valuable biological information and enable basecaller optimization and modification detection. These raw signals also underpin algorithms that could improve ONT device portability and enhance target enrichment efficiency through adaptive sampling. Nevertheless, the storage and sharing of raw nanopore data remain limited due to technical constraints and the lack of standardized and centralized infrastructure. To address this challenge, we developed SquiDBase (https://squidbase.org), a dedicated repository for raw microbial nanopore sequencing data with linked processed data and metadata. To maximize immediate utility, we built SquiDPipe, a Nextflow pipeline for the automated removal of human reads from raw nanopore data, sequenced 24 clinically relevant viruses and incorporated them into SquiDBase, and added publicly available reference datasets and new community contributions. By offering a centralized, open-access raw data collection platform, SquiDBase facilitates data sharing, enhances reproducibility, and supports the development and benchmarking of computational tools, reinforcing open science in nanopore sequencing.
|
|
Scooped by
mhryu@live.com
Today, 12:37 AM
|
Precise modulation of gene expression via cis-regulatory editing holds promise for non-transgenic crop improvement, but the sequence-to-function relationships that govern plant promoter activity remain poorly understood. Here, we develop a massively parallel reporter assay (MPRA) in Sorghum bicolor to systematically measure the effects of >30,000 CRISPR-like mutations- deletions, substitutions, and motif insertions- across entire native promoters and 5′ untranslated regions (UTRs) of three photosynthesis genes: PsbS, Raf1, and SBPase. We find that gene expression is most tunable within a ~500 base pair core promoter region, where mutational effects are reproducible across biological replicates and predictive of protein output. Within these regions, we identify compact deletions and motif insertions that strongly increase protein production (>30-fold relative to wild type), exceeding the performance of transgenic enhancer elements. Mutation-effect relationships are gene-specific, highlighting the need for tailored regulatory maps. Our results establish a high-throughput strategy for cis-regulatory fine-mapping that enables crop improvements via minimal, precise, and non-transgenic gene edits.
|
|
Scooped by
mhryu@live.com
January 8, 11:30 PM
|
Species barriers limit gene flow and maintain co-adapted genomes. Interspecific hybridization can break down species barriers to reveal genetic incompatibilities. Although the phenotypic and genomic consequences of hybridization have been extensively studied in many systems, far less is known about the longer-term evolutionary dynamics of highly divergent, mosaic genomes and the extent to which genetic incompatibilities shape their adaptation. In yeast, pre-zygotic barriers are weak but post-zygotic barriers are strong due to mispairing of chromosomes during meiosis. By suppressing anti-recombination genes in meiosis, we generated a panel of 20 haploid recombinant hybrids from a cross between Saccharomyces cerevisiae and its sister species, Saccharomyces paradoxus. Across conditions, these hybrids are, on average, less fit than either parent and show broad phenotypic variation. Inheritance patterns of protein complexes in the hybrid genomes reveal no evidence of pairwise lethality but do support a model of pervasive weak negative genetic interactions in hybrid protein complexes. We show by laboratory evolution that each recombinant genome follows a distinct evolutionary trajectory, and a small subset of hybrid protein complexes and loci show hybrid-specific mutational targeting. Finally, we show that species-of-origin alleles can bias evolutionary outcomes by reshaping selection on interacting genes. Together, our results suggest that strong pairwise incompatibilities are rare, while weak, background-dependent incompatibilities are widespread and shape fitness and adaptation in hybrid genomes.
|
|
Scooped by
mhryu@live.com
January 8, 10:59 PM
|
Computational workflows for MS-based proteomics remain comparatively fragmented, with heterogeneous data formats and analysis pipelines that hinder their reproducibility, interoperability, and reuse of processed data. We present msmu, an open-source Python package that implements a flexible and reproducible end-to-end pipeline for post-search data preprocessing and statistical analysis. At its core, msmu leverages the highly structured MuData format, empowering comprehensive data provenance, transparency in data sharing and reuse, and interoperability with broader Python ecosystem. Together, msmu represents a unique and significant step toward realizing the FAIR (Findable, Accessible, Interoperable, and Reusable) principles in computational proteomics.
|
|
Scooped by
mhryu@live.com
January 8, 10:40 PM
|
Cold seeps host diverse microbes and viruses with numerous unexplored defense and anti-defense systems. Analysis of 3813 microbial and 13,336 viral genomes from 191 metagenomes across 17 cold seep sites reveals extensive microbial defense repertoires, with over 60% representing candidate systems. Experimental validation confirms that several candidates protect against viral infection. These defense systems frequently co-occur, suggesting potential synergistic interactions, and are broadly distributed across sediments. In response, viruses have evolved diverse anti-defense genes, and the concurrent presence of multiple viral and microbial systems highlights intricate coevolution. Functionally critical lineages, such as anaerobic methanotrophic archaea, sulfate-reducing bacteria, and diazotrophs, appear to modify their defensive strategies under ecological and environmental pressures; for example, sulfate-reducing bacteria harbor multiple Gabija systems while corresponding viruses carry anti-Gabija genes, illustrating specific coevolutionary adaptations. Overall, these findings underscore the critical role of virus-microbe interactions in shaping microbial metabolic functions and environmental adaptation in deep-sea ecosystems. Deep-sea cold seeps harbor diverse microbial and viral communities engaged in molecular conflicts. This study reveals extensive and coordinated defense and anti-defense systems that coevolve and shape microbial–virus relationships in this extreme environment.
|
|
Scooped by
mhryu@live.com
January 8, 7:40 PM
|
The messenger RNA 3' untranslated region contains important regulatory sequences, including upstream sequence elements (USEs), which regulate gene expression. One well-characterised USE in the 3'UTR of the Drosophila polo gene affects adult fly phenotypes when disrupted. We have now identified a highly conserved sequence within this USE (DplUSE) in the 3'UTR of several vertebrate genes, including in zebrafish, mouse, and human genomes and show that DplUSE enhances gene expression in human cells and zebrafish embryos. We show that, in humans, DplUSE-containing genes are associated with congenital disease processes, and that disruption of DplUSE function impairs zebrafish development. We also found that HuR/ELAVL1, hnRNPC, and PTBP1/hnRNPI bind to DplUSE RNA and are required for its activity in a human cell line, suggesting a highly conserved mechanism across distantly related species. Our results indicate that PTBP1 has a global function in alternative polyadenylation, activating the selection of distal polyA sites and repressing intronic polyadenylation in DplUSE-containing genes while hnRNPC and HuR modulate their expression. Additionally, we found that a colon cancer-associated SNP in the POU2AF2/C11orf53 3'UTR creates an ectopic DplUSE site, increasing gene expression in zebrafish gut cells and in a human cell line. We have therefore identified a short 3'UTR motif present in diverse vertebrate genes that controls their expression through conserved RBPs interactions and is implicated in human disease.
|
|
Scooped by
mhryu@live.com
January 8, 7:24 PM
|
The expansion of the genetic alphabet through the development of unnatural base pairs (UBPs) has the potential to revolutionize synthetic biology and biotechnology. However, the replication of the UBPs by eukaryotic DNA polymerases is largely unexplored and the sequencing of the UBPs remains challenging. Herein, we explored and demonstrated the activity of human DNA polymerase β (Pol β) for the efficient and specific synthesis and extension of a panel of representative UBPs, including dNaM–dTPT3, dCNMO–dTPT3, and their functionalized derivatives. Based on this, we established a method for the sequencing of DNAs containing different unnatural bases, involving stalled primer extension mediated by Pol β, selective conversion of an unnatural nucleotide into two different natural ones in parallel and further primer extension mediated by Taq DNA polymerase, and Sanger or deep sequencing of the produced natural DNAs to locate the unnatural bases. The precision, universality, and potential for high-throughput applications of this method were demonstrated by the successful sequencing of various DNAs containing one or multiple of different unnatural bases. This work suggests the possibility of integrating the UBPs into the eukaryotic DNA replication systems and provides a technical foundation for the robust sequencing of DNAs with an expanded genetic alphabet.
|
|
Scooped by
mhryu@live.com
January 8, 7:14 PM
|
The KEGG Orthology (KO) system links DNA and protein sequences to biological functions and pathways, providing a curated, fundamental and consistent annotation framework across all domains of life. While accurate, traditional sequence alignment-based annotation methods are computationally expensive, which severely limits their application in large-scale datasets. To address this challenge, we introduce DeepKOALA, a deep learning approach based on Gated Recurrent Units (GRU), which frames KO annotation as an open-set recognition task. This design reduces false positives arising from out-of-scope sequences and, together with a lightweight GRU backbone, enables high-throughput annotation. The GRU-based model was benchmarked against four other deep learning architectures and showed the best balance between speed and accuracy. We further performed a cross-species evaluation of DeepKOALA in comparison with existing KO annotation tools, where it achieved an F1-score of 0.8653 and 37.5-fold acceleration compared with BlastKOALA. We also provide a specialized fragment model for handling incomplete sequences and an optional Multi-domain mode. Together, these features make DeepKOALA a scalable, efficient, and accurate solution for high-throughput function annotation.
|
|
Scooped by
mhryu@live.com
January 8, 5:36 PM
|
Nitazenes are an emergent class of synthetic opioids that often rival or exceed fentanyl in their potency. These compounds have been detected internationally in illicit drugs and are the cause of increasing numbers of hospitalizations and overdoses. New analogs are consistently released, making detection challenging — new ways of testing a wide range of nitazenes and their metabolic products are urgently needed. Here, we develop a computational protocol to redesign the plant abscisic acid receptor PYR1 to bind diverse nitazenes and maintain its dynamic transduction mechanism. The best design has a low nanomolar limit of detection in vitro against nitazene and menitazene. Deep mutational scanning yielded sensors able to recognize a range of clinically relevant nitazenes and the common metabolic byproduct in a complex biological matrix with limited cross-specificity against unrelated opioids. Application of protein design tools on privileged receptors like PYR1 may yield general sensors for a wide range of applications in vitro and in vivo. Nitazenes are potent synthetic opioids that are difficult to detect. Here, authors computationally redesign a plant receptor to create sensitive sensors capable of detecting diverse nitazenes and their metabolites in biological samples.
|
|
Scooped by
mhryu@live.com
January 8, 5:30 PM
|
CRISPR/Cas-based genome editing has revolutionized plant biotechnology, enabling precise genomic modifications for crop improvement and functional genomics. The success of these applications hinges on designing single guide RNAs (sgRNAs) that maximize on-target efficiency while minimizing off-target effects. Current resources for sgRNA design and performance evaluation in plants are fragmented and lack integration with genomic and epigenomic context, which influences both editing efficacy and specificity. Here, we present PCdb (Plant CRISPR Database; https://gmo.sjtu.edu.cn/pcdb), a comprehensive plant-focused database by integrating experimentally validated sgRNAs, their annotated genomic contexts, genome-wide off-target predictions, and multi-layered epigenomic annotations. PCdb encompasses 6,172 manually curated editing records from 2,132 publications, covering 4,320 unique sgRNAs and 6,117,424 predicted off-target sites across nine major plant species. Uniquely, PCdb contextualizes potential editing outcomes-both on-target and off-target-within the chromatin landscape by incorporating DNA methylation profiles, chromatin accessibility data, and histone modification patterns. The database features an intuitive web interface supporting flexible queries, interactive visualization tools, and comprehensive analytical modules for both sgRNA efficiency assessment and off-target analysis. A case study reanalysis of Oryza sativa yield-related genes demonstrates PCdb's capability to provide a comprehensive performance profile, evaluating both on-target characteristics and off-target risks within their native epigenomic context. Through systematic analysis of the database, we reveal critical sequence and chromatin features influencing editing outcomes, providing novel insights for improved gene editing efficacy and specificity.
|
|
Scooped by
mhryu@live.com
January 8, 4:49 PM
|
Temperate phage can transmit both horizontally (lytic cycle) and vertically (lysogenic cycle). Many temperate phage have the ability to modify their lysis/lysogeny decisions based on various environmental cues. For instance, many prophage are known to reactivate when SOS stress responses of their host are triggered. Temperate phage infecting Bacilli can also use peptide signals (“arbitrium”) to control their lysis/lysogeny decisions. However, information from the arbitrium and SOS systems can be potentially conflicting, and it is unclear how phage integrate information carried by these two different signals when making lysis–lysogeny decisions. Here, we use evolutionary epidemiology theory to explore how phage could evolve to use both systems to modulate lysis/lysogeny decisions in a fluctuating environment. Our model predicts that it can be adaptive for phage to respond to both host SOS systems and arbitrium signaling, as they provide complementary information on the quality of the infected host and the availability of alternative hosts. Using the phage phi3T and its host Bacillus subtilis, we show that during lytic infection and as prophage, lysis–lysogeny decisions rely on the integration of information on host condition and arbitrium signal concentrations. For example, free-phage are more likely to lysogenise a stressed host, and prophage are less likely to abandon a stressed host, when high arbitrium concentrations suggest susceptible hosts are unavailable. These experimental results are consistent with our theoretical predictions and demonstrate that phage can evolve plastic life-history strategies to adjust their infection dynamics to account for both the within-host environment (host quality) and the external environment that exists outside of their host (availability of susceptible hosts in the population). More generally, our work yields a new theoretical framework to study the evolution of viral plasticity under the influence of multiple environmental cues.
|
|
|
Scooped by
mhryu@live.com
Today, 1:07 AM
|
Aspergillus niger serves as a cell factory for efficient enzyme production. The lipase derived from Thermomyces lanuginosus (TLL) is known for its remarkable thermal stability and is extensively utilized in various industrial fields. In this study, a heterologous expression strain, ΔAnTll-11, of TLL was successfully constructed in A. niger. Through fermentation optimization, the lipase activity was enhanced 8.7-fold to 4547.95 U/mL, compared with the initial value of 520 U/mL. Enzymatic characterization indicated that the recombinant lipase exhibited optimum activity at pH 9.5 and 45 °C, and its activity was positively influenced by Ca2⁺, Ag⁺, Mg2⁺, and Cu2⁺. The ΔAnTll-11 strain exhibited enhanced tolerance to high osmolarity, oxidative stress, and thermal shock. Furthermore, transcriptomic analysis of the ΔAnTll-11 strain revealed that half of the total annotated genes (7199 genes) were differentially expressed genes (DEGs). According to protein‒protein interaction network and weighted gene coexpression network analyses, genes involved in ribosome function, amino acid metabolism, glycosylation, energy metabolism, and the MAPK signalling pathway may affect the expression of lipase. The transcriptomic findings elucidated the regulatory mechanisms by which A. niger expresses foreign proteins and enzymes, establishing a groundwork for further enhancing A. niger as a cell factory for efficient enzyme and protein production.
|
|
Scooped by
mhryu@live.com
Today, 12:59 AM
|
The global burden of antimicrobial resistance demands the urgent development of new antibiotics. To face this threat, Torres et al. leveraged deep-learning models to identify archaeal encrypted peptides (termed ‘archaeasins’) with potential antimicrobial activity. This work highlights Archaea as an underexplored but potentially rich source of antibiotics.
|
|
Scooped by
mhryu@live.com
Today, 12:53 AM
|
Paclitaxel, a clinically potent anticancer drug derived from Taxus species, faces persistent challenges in sustainable supply. Synthetic biology presents substantial opportunities for its de novo production, particularly with recent breakthroughs in elucidating its intricate biosynthetic pathways. However, its heterologous biosynthesis is significantly constrained by key bottlenecks, including pathway complexity, poor P450 expression, and inefficient metabolic flux. In this study, we explore how synthetic biology facilitates pathway decoding and reconstruction and propose strategies involving nonclassical chassis such as plant-associated cyanobacteria and filamentous fungi to enhance P450 compatibility. We also present a pragmatic framework for the rational application of state-of-the-art tools, including cell-free systems, synthetic microbial consortia, hybrid chemoenzymatic synthesis, and machine learning, to sustainably produce paclitaxel and other natural products.
|
|
Scooped by
mhryu@live.com
January 8, 11:34 PM
|
The boom of microbiome research in agriculture over the past several decades allows scientists, growers, policymakers, and businesses to collaborate on a unique opportunity—deploying microbiomes and microbiome attributes for the improvement of crop production. The idea of translational microbiomes is well established in the medical field; however, this framework is relatively new to agriculture. In this review, we discuss a series of methodologies grounded in microbiome science to enhance crop health. These include diagnostic approaches (pathogen and toxin detection and the monitoring of stress-related community ecology patterns) and intervention strategies (synthetic communities, microbiome-aware crop management practices, passaging microbiomes, and exploiting the vertical and lateral transmission of microbiomes to seeds). Developing and implementing these approaches remain challenging due, in part, to a shortage of long-term in situ studies demonstrating the robustness and effectiveness of translational microbiome efforts against the background of heterogeneity and ecological complexity of agricultural systems. Moreover, the cost and availability of ‘omics methods central to microbiome analysis, disparate standards for microbiome product development, and limited longstanding relationships with stakeholders have slowed down the application of microbiome-based solutions. However, the increasing cost-effectiveness of microbiome approaches in crop management makes translational microbiomes likely assets in the movement toward precision agriculture. This “personalized treatment” for plants holds promise for improved food security and environmental sustainability, by reducing commonplace synthetic amendments and promoting native microbial biodiversity.
|
|
Scooped by
mhryu@live.com
January 8, 11:11 PM
|
Microbial metabolism relies on redox reactions that exploit chemical disequilibria. While aerobic carbon oxidation, carbon fixation, and fermentation are well studied, the broader space of anaerobic carbon redox reactions remains underexplored. In this study, carbon comproportionation, or reverse fermentation, reactions are identified as a previously unrecognized and potentially favorable class of microbial carbon redox transformations. Particular attention is given to the reaction between methane (CH4) and carbon monoxide (CO) to form acetate, a reaction that has not previously been evaluated despite the widespread occurrence of CH4 and CO in anoxic systems. Gibbs energies (ΔGr) for this reaction were calculated across broad ranges of temperature, pH, and dissolved CH4 and CO concentrations using measured physicochemical data from a wide variety of environmental systems. We show that acetogenic CH4-CO comproportionation is exergonic in all environments where both substrates were detected. The most favorable energetic conditions occur at high pH, low temperature, and high reactant concentrations, consistent with cool serpentinizing systems. In several settings, the calculated Gibbs energy yields and energy densities overlap or exceed known anaerobic metabolisms involving CH4, CO, and acetate. These results demonstrate that acetogenic CH4-CO comproportionation can support microbial energy conservation in a variety of settings. To determine if this metabolism could have operated on early Earth or Mars, modeled fluid compositions show that this reaction is also exergonic under plausible physicochemical regimes. This work broadens the suite of possible microbial energy metabolisms and provides testable criteria for evaluating carbon-based catabolic reactions on Earth and on other planetary bodies.
|
|
Scooped by
mhryu@live.com
January 8, 10:52 PM
|
The adaptation of the CRISPR/Cas system as a biotechnological tool has enabled a wide spectrum of targeted genome modifications. Whereas earlier approaches focused on small sequence changes, recent years have seen a shift toward larger-scale alterations. Advances in homology-directed gene targeting now enable efficient, scar-free kilobase insertions, while combining nuclease-deficient Cas effectors with recombinases or transposases allows the integration of much larger sequences. Prime editing further expands this scope, enabling inversions, replacements, and deletions spanning hundreds of kilobases to several megabases. More recently, genome engineering has reached a new stage with chromosome fission and fusion, demonstrating the feasibility of controlled karyotype restructuring. Together, these advances open new opportunities for crop improvement, from establishing reproductive barriers and mimicking evolutionary processes to trait stacking on Plant Artificial Chromosomes.
|
|
Scooped by
mhryu@live.com
January 8, 7:46 PM
|
Bacterial RNA polymerase binds and unwinds promoter DNA to initiate transcription. The process is often inefficient and can be stimulated by activator proteins. For example, many activators bind RNA polymerase and promoters simultaneously, stabilizing their interaction. Working with the multiple antibiotic resistance activator (MarA) protein of E. coli, we have discovered an alternative mechanism. We show that, when bound upstream of the flgBCDEFGHIJ/KL operon, MarA perturbs base pairing adjacent to its DNA target. This compensates for inefficient duplex unwinding by RNA polymerase and, as a result, activates transcription. Consistent with our model, an appropriate base pair mismatch mimics the effect of, and removes the need for, MarA. As many regulators alter DNA conformation, we suggest that this mechanism of activation could be commonplace.
|
|
Scooped by
mhryu@live.com
January 8, 7:36 PM
|
Precise modeling of transcriptional regulation is essential for the rational design of genetic circuits in synthetic biology. Current computational approaches for predicting transcriptional activity (ITX) typically lack mechanistic clarity, composability, and scalability, and require extensive training data. Here, we present a modular thermodynamic modeling framework that explicitly parameterizes molecular interactions among promoters, RNA polymerase (RNAP) and transcription factors (TFs). Implemented as the computational platform, T-Pro, this approach provides robust interpretability, scalability, and predictive power. Experimental validation across three distinct bacteria—Escherichia coli, Bacillus subtilis, and Corynebacterium glutamicum—demonstrates substantial improvements (up to 20-fold) in a composite transcriptional performance metric (Fmax*FC), achieved within only three Design–Build–Test–Learn cycles and fewer than five genetic constructs in total. Furthermore, we validate the framework by engineering multispecies bacterial communication circuit, highlighting its broad utility and generalizability. The principles and tools developed here thus enable efficient, rational optimization of transcriptional regulation across diverse prokaryotic hosts.
|
|
Scooped by
mhryu@live.com
January 8, 7:18 PM
|
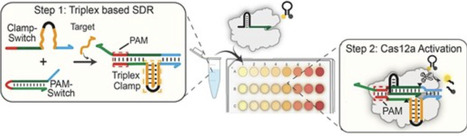
We present a molecular strategy that enables the programmable activation of the CRISPR–Cas12a system in response to triplex DNA formation triggered by single-stranded DNA (ssDNA) or RNA inputs. Our triplex-controlled Cas12a assay leverages the high specificity of clamp-like triplex structures to control a toehold-based strand displacement reaction within a rationally designed DNA hairpin (PAM-Switch). Upon displacement and protospacer adjacent motif (PAM) complementation, the Cas12a ribonucleoprotein (RNP) is activated, initiating trans-cleavage and producing a concentration-dependent fluorescent signal. By decoupling target recognition (via triplex formation) from direct hybridization with the Cas12a–crRNA complex, the assay eliminates the need for target-specific crRNAs. This design also allows multiple detection of distinct nucleic acid (NA) targets using a single Cas12a reaction mix. Through the use of triplex-based clamps, the proposed platform achieves enhanced specificity for single-nucleotide variants and supports the detection of both ssDNA and RNA targets across a broad range of lengths (10–20 nucleotides), addressing key limitations in current Cas12a-based diagnostics and opening new avenues for NA sensing.
|
|
Scooped by
mhryu@live.com
January 8, 7:11 PM
|
The Encyclopedia of Domains (TED) provides domain annotations for proteins in the AlphaFold Protein Structure Database (AFDB) using a consensus of three state-of-the-art structure-based methods. We used these TED domain annotations to construct profile Hidden Markov models (HMMs), collectively forming the TED Library of HMM (TEDLH). TEDLH enables sensitive sequence and profile searches, supporting systematic exploration of protein domain families and their evolutionary relationships. TEDLH links domain HMMs to experimentally determined CATH-PDB structures through direct (primary) and transitive (secondary and tertiary) relationships. Fewer than half of TEDLH HMMs are directly linked to a CATH-PDB domain; the remaining models are connected through transitive relationships. These transitive links extend coverage into more divergent regions of sequence space and better represent CATH superfamily diversity. HMM-HMM comparisons within CATH superfamily 3.30.70.100 illustrate how transitive relationships expand sequence coverage in TEDLH. In this superfamily, 4,813 TEDLH HMMs are connected to 212 CATH-PDB representatives. Primary, secondary, and tertiary relationships progressively capture more divergent sequences (pairwise sequence identity <20%) that retain structural similarity (TM-score >0.6) and a conserved two-layer α/β sandwich core fold. All-against-all HMM-HMM comparisons across TEDLH also reveal sequence similarities across the CATH hierarchy (cross-hits). At low query coverage (<50%), cross-hits are more frequent between CATH classes, whereas at higher coverage thresholds (>70%) they predominantly occur between superfamilies. These cross-hits are not driven by superfamily size or sequence diversity and can provide guidance for CATH curation. As an example, analysis of cross-hits between superfamilies 2.170.130.30 and 3.10.20.30 reveals evolutionary relationships between these groups.
|
|
Scooped by
mhryu@live.com
January 8, 5:35 PM
|
Antifungal resistance is a growing crisis affecting human health and agriculture that could be accelerated by global change. We call for a One Health research agenda to systematically investigate environmental drivers and inform policy to mitigate the potential of global environmental change in fuelling antifungal resistance.
|
|
Scooped by
mhryu@live.com
January 8, 4:57 PM
|
Metabolites generated by host and pathogen have a major impact on the severity and outcomes of infection. The metabolic response to infection shapes the nature and intensity of the immune response, both in bloodstream infections and, especially, in the pathogenesis of pneumonia. Some metabolites are closely linked to pro-inflammatory responses, whereas others act as immunomodulators in mitigating damage to the host, a common consequence of inflammation. Immunometabolites are also major factors in driving bacterial adaptation to the host, enabling pathogens acquired from environmental sources to modify their gene expression to optimize for persistent infection. In this era of diminishing antimicrobial efficacy, an appreciation of the immunometabolic responses to bacterial infection may provide novel targets for therapy.
|