Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 5:43 PM
|
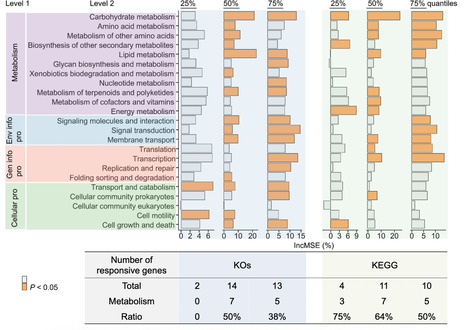
Microbes, as the planet’s most abundant and diverse organisms, drive soil functions globally and are vulnerable to environmental stressors triggered by global change. Yet, knowledge regarding the impacts of multiple environmental stressors on their functional profiles as well as the consequences for soil functionality largely remains unknown. Here, we analyze two global-scale datasets including information on soil metagenomics and multiple environmental stressors. We find that across terrestrial ecosystems worldwide, up to 60% of all functional genes significantly shift when soil microbes experience the high-level of concurrent stressors. In this regard, the relative abundances of genes involved in microbial growth are negatively linked to the increasing number of stressors. Conversely, those genes linked to stress resistance and energy production exhibit positive responses. Taken together, our findings highlight a significant restructuring of global soil functional microbiomes in response to multiple environmental stressors. Consequently, such restructuring drives community-level shifts in matter and energy reallocations, thereby impacting the maintenance of soil functionality under the projected global change. Soil microbes drive ecosystem functions but are vulnerable to environmental stressors triggered by global change. This study reveals that multiple environmental stressors drive community-level restructuring of soil functional microbiomes globally.
|
|
Scooped by
mhryu@live.com
Today, 2:54 PM
|
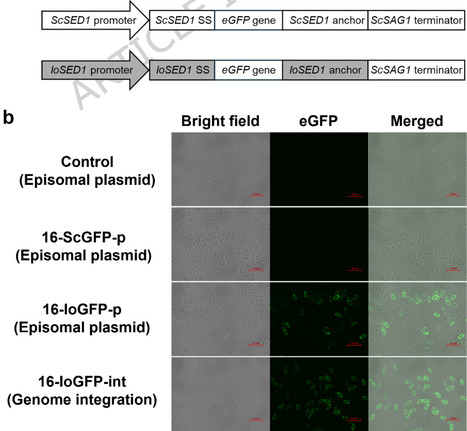
Displaying cellulolytic enzymes on the yeast cell surface via the anchoring domain of glycosylphosphatidylinositol-anchored proteins can integrate enzyme production, saccharification, and fermentation into a single step in biomass fermentation. Therefore, this technique represents a promising strategy for cost-effective and sustainable production of value-added chemicals and biofuels from lignocellulosic biomass (LCB), the most abundant renewable resource, which typically requires multiple complex processing steps. However, pretreatment of LCB, such as acidic thermochemical treatment, causes harsh cultivation conditions characterized by low pH, high temperature, and lignocellulosic fermentation inhibitors. Thus, host strains displaying cellulolytic enzymes must exhibit remarkable tolerance to these stresses. Here, we employed the non-conventional yeast Issatchenkia orientalis, which has remarkable multi-stress tolerance, and successfully established the display system in this yeast. We found that the cell-surface display cassette that functions in the model yeast Saccharomyces cerevisiae was not functional in I. orientalis; therefore, we constructed a modified display cassette based on the I. orientalis SED1 gene. Using this cassette, we successfully displayed fluorescent protein and β-glucosidase (BGL) on the cell surface. The BGL-displaying strain grew robustly on cellobiose as the sole carbon source and, notably, maintained growth even in the presence of lignocellulosic fermentation inhibitors. To the best of our knowledge, this is the first report demonstrating the successful immobilization of functional proteins on the cell surface of I. orientalis and assimilation of LCB-derived intermediates, even under stress conditions, by displaying cellulolytic enzymes, highlighting its potential as a general industrial platform for sustainable LCB bioconversion. Key points A cell-surface display cassette for Issatchenkia orientalis was constructed.The β-glucosidase-display strain utilized cellobiose as the sole carbon source.Cellobiose assimilation was observed even in the presence of fermentation inhibitors.
|
|
Scooped by
mhryu@live.com
Today, 12:48 PM
|
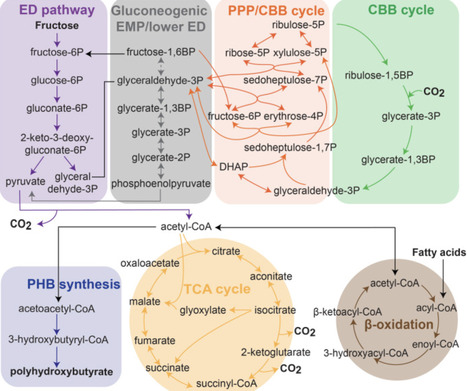
Cupriavidus necator strain H16 has emerged as a versatile microbial chassis for sustainable bioproduction due to its metabolic flexibility, enabling growth on a wide range of substrates, including H2 + CO2, formate, and organic carbon (waste) sources such as volatile fatty acids. This review first provides a comprehensive overview of recent advances in systems-level understanding and metabolic engineering of C. necator. We next discuss recent advances in omics analyses and genome-scale metabolic modeling that are increasingly used to understand the large genome and wide metabolic portfolio of C. necator. We further discuss the native metabolic network, including autotrophic growth via the Calvin Benson Bassham (CBB) cycle, as well as heterotrophic and mixotrophic capabilities. Engineering strategies to enhance substrate utilization and conversion efficiency particularly for H2 + CO2, formate, and mixed feedstocks are discussed alongside efforts to expand the substrate range in this organism. Other than the already industrialized production of the bioplastic polyhydroxybutyrate (PHB) and related polyhydroxyalkanoates in C. necator we provide an overview of the wide range of products for which proof-of-principles have been shown in C. necator. We also discuss recent advances in bioprocess design for gas fermentation, electromicrobial production, and H2-based biocatalytic reduction using C. necator. Finally, we compare C. necator with other hydrogen and formate-utilizing bacteria to identify key knowledge gaps and outline future directions for advancing C. necator as a host for sustainable industrial biotechnology.
|
|
Scooped by
mhryu@live.com
Today, 11:57 AM
|
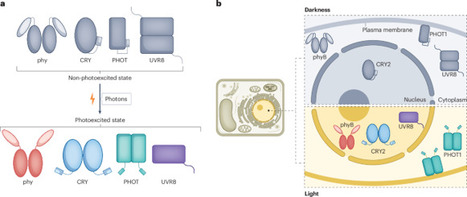
Plants decode environmental light cues through photoreceptors to orchestrate growth and developmental programs. Upon photon absorption, photoreceptor proteins transition from non-photoexcited to photoexcited states, with most functional studies focusing on the activities of the photoexcited form. However, emerging evidence reveals that non-photoexcited photoreceptors exert distinct biological functions in darkness, suggesting unexpected complexity in photon-mediated signalling. In this Perspective, we enumerate current observations underpinning photoreceptor-mediated control of plant development in darkness, categorize mechanisms of photon-dependent modulation and propose an expanded conceptual model that integrates both photoexcited and non-photoexcited activities. These insights reassess the previously overlooked importance of darkness, redefining light and darkness as dynamic, multidimensional regulators of plant physiology. In a manner distinct from their light-activated canonical roles, non-photoexcited photoreceptors can also exert biological functions in darkness, suggesting unexpected complexity in photon-mediated signalling.
|
|
Scooped by
mhryu@live.com
Today, 11:32 AM
|
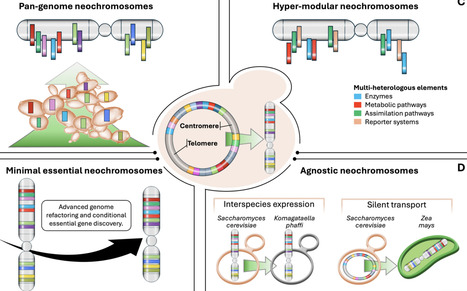
The evolution of the yeast, Saccharomyces cerevisiae, from a genetically tractable model organism to a chassis for genome-scale engineering represents one of the most influential trajectories in eukaryotic biology. The Synthetic Yeast Genome Project (Sc2.0) embodies the current height of this trajectory, having now delivered functional synthetic versions of all 16 native yeast chromosomes and bringing the construction of the first fully synthetic eukaryotic cell within reach. Beyond its technical achievements, Sc2.0 has reshaped how eukaryotic genomes are understood and explored through iterative design-build-test-learn (DBTL) cycles, and reframed the yeast genome as a dynamic, highly modifiable system rather than a static biological blueprint. Moreover, the progress on genome engineering pipelines and synthetic biology has laid the foundations for the de novo development of modular synthetic chromosomes (neochromosomes) that operate orthogonally to the native genome. These synthetic platforms provide dedicated, large-scale genomic landing pads for refactoring genetic networks, reallocating redundancy, and introducing large, multiplexed gene assemblies, thereby extending yeast engineering toward programmable and hyper-versatile biological systems. To commemorate the 40th anniversary of the journal Yeast, this mini-review celebrates the exceptional power of yeast genetics, outlining key conceptual and technological advances emerging from the Sc2.0 endeavour and beyond. Finally, we examine the cross-cutting engineering insights and the future potential of neochromosomes for the next generation of synthetic yeasts.
|
|
Scooped by
mhryu@live.com
Today, 11:17 AM
|
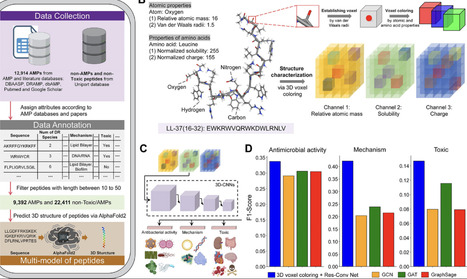
Artificial intelligence (AI)-driven discovery of antimicrobial peptides (AMP) is yet to fully utilize their three-dimensional (3D) structural characteristics, microbial species-specific antimicrobial activities, and mechanisms. Here, we constructed a QLAPD database comprising the sequence, structures, and antimicrobial properties of 12,914 AMPs. QLAPD underlies a multimodal, multitask, multilabel, and conditionally controlled AMP discovery (M3-CAD) pipeline, proposed for the de novo design of multi-mechanism AMPs to combat multidrug-resistant organisms (MDROs). This pipeline integrates generation, regression, and classification modules, using an innovative 3D voxel coloring method to capture the nuanced physicochemical context of amino acids, thus enhancing structural characterizations. QLX-3DV-1 and QLX-3DV-2, identified through M3-CAD, were found to demonstrate multiple antimicrobial mechanisms, notable activity against MDROs, and low toxicity. In vivo experiments were used to validate their antimicrobial effects with limited local and systemic toxicity. Overall, integrating 3D features, species-specific antimicrobial activities, and mechanisms enhanced AI-driven AMP discovery, making the M3-CAD pipeline a viable tool for de novo AMP design.
|
|
Scooped by
mhryu@live.com
Today, 10:58 AM
|
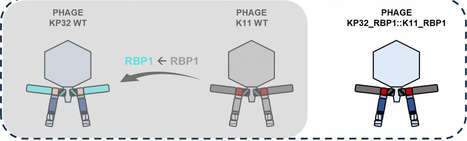
Branched receptor-binding protein (RBP) systems enable bacteriophages to broaden their host range through the incorporation of two or more RBPs. Using the Klebsiella podophages KP32, K11, and KP34 as model systems, we experimentally validated the interaction between the branching domain of the primary RBP (RBP1) and the conserved docking peptide of the secondary RBP (RBP2) as an essential architectural pair enabling dual-RBP incorporation into the virion. Systematic engineering revealed that loss of either of these domains, the branching domain or the conserved peptide, abolishes RBP2 assembly, underscoring their structural role in organizing the branched configuration and demonstrating that the anchor domain is the sole element directly attaching the RBP complex to the virion. Exploiting this interaction, we engineered a chimeric phage based on the Klebsiella phage KP32 scaffold that was capable of cross-genus infection and productive propagation on both Klebsiella and Escherichia hosts. In contrast to previous approaches that required replacement of the entire RBP, the adaptor and nozzle proteins, this strategy achieved host-range reprogramming through modular domain swapping and positional relocation of RBPs (i.e., exchanging RBP1 and RBP2 positions). Conversely, an Escherichia phage K1F scaffold was also successfully engineered to infect Klebsiella. Our study confirms that the RBP branching domain and the conserved peptide function as specific interacting partners. Our findings establish the conserved peptide as a docking element and highlight the structural flexibility of podoviruses to accommodate RBPs from different positional and taxonomic contexts. Collectively, this work provides a mechanistic framework for rational phage engineering and defines a general design principle for generating customized therapeutic phages with an expanded host spectrum, including cross-genus infectivity.
|
|
Scooped by
mhryu@live.com
Today, 2:08 AM
|
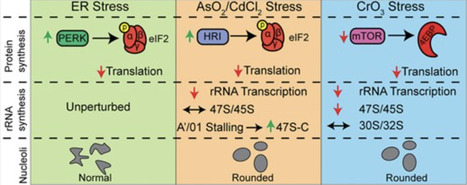
Ribosome synthesis is one of the most energy-intensive processes in a growing cell, consuming >60% of cellular energy reserves. As such, ribosome biogenesis is highly sensitive to stress to prevent costly expenditures under adverse conditions. Moreover, successful assembly requires precise stoichiometric balance between ribosomal proteins and ribosomal RNAs (rRNA). Here, we define novel regulatory mechanisms of ribosome biogenesis under stress that reveal previously unrecognized aspects of rRNA maturation. We demonstrate that early pre-rRNA processing is particularly sensitive to stress induced by environmentally relevant heavy metals. Surprisingly, our analysis shows that 5′ and 3′ end processing can be uncoupled in human cells, with 3′ end cleavage occurring independently of 5′ end processing. We further show that classical inducers of endoplasmic reticulum stress suppress ribosomal protein synthesis without inhibiting rRNA transcription, leading to an imbalance between these essential components of ribosome assembly. This imbalance may exacerbate cellular stress and compromise proteostasis. Together, our findings uncover stress-specific checkpoints in ribosome biogenesis that link environmental exposures to disrupted nucleolar function and highlight new layers of regulation in human rRNA maturation.
|
|
Scooped by
mhryu@live.com
Today, 1:57 AM
|
Bioorthogonal noncanonical amino acid tagging (BONCAT) bridges the RNA-informed translatome and proteome by isolating newly synthesized proteins. Recent implementation in plants has demonstrated the potential of BONCAT experimentation, which still lags behind its utilization in other fields. In this forum, current and potential future uses of BONCAT in plants are discussed.
|
|
Scooped by
mhryu@live.com
Today, 1:50 AM
|
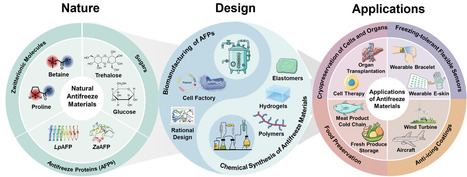
Ice formation poses significant challenges across multiple domains, including biomedicine, food industry, infrastructure, and intelligent sensors, where freezing environments can cause serious functional and safety issues. The development of effective antifreeze materials has become an urgent priority. Nature offers valuable insights in this regard, having evolved diverse psychrotolerant organisms from microorganisms to plants and fish. Within these organisms, key small molecules and macromolecules responsible for cold tolerance have been progressively identified. Inspired by them, recent years have witnessed the design and synthesis of a series of high-performance antifreeze materials through biomanufacturing or chemical synthesis. This review highlights the significant progress in antifreeze materials, tracing their evolution from natural models to rational design systems: (1) natural antifreeze materials and their mechanistic insights, with emphasis on molecular lessons for ice inhibition; (2) biomanufacturing and rational design of antifreeze proteins based on emerging structure-activity relationships; (3) nature-inspired synthetic antifreeze materials, such as polymers, hydrogels, and elastomers; and (4) key applications in cryopreservation, food preservation, anti-icing coatings, and freezing-tolerant flexible sensors. While promising advances have been made, this review also addresses persistent challenges in translating these laboratory innovations into scalable applications.
|
|
Scooped by
mhryu@live.com
Today, 1:33 AM
|
Cells can respond to alterations in the abundances of specific proteins through transcriptional outputs. Synthetic approaches inspired by native post-transcriptional circuits that convert protein abundance changes into programmable gene expression would be transformative. Here, we discover and describe design principles that effectively convert protein degradation into transcriptional outputs in live cells. We define ratiometric transcriptional activation, where control over the ratio between a transcriptional inhibitor-protein of interest fusion and transcription factor enables detection of abundance changes with high sensitivity at scale. We show that ratiometric transcriptional activation can be implemented in single cells using triply orthogonal circuits or in multicellular pools, operating independently of mechanism of protein downregulation and enabling simultaneous detection of multiple protein downregulation events through outputs such as cell survival, fluorescent protein expression, or barcode sequencing. These circuits can be applied to oncogenic targets and enable discovery of new molecular glue degraders.
|
|
Scooped by
mhryu@live.com
Today, 1:11 AM
|
Biological systems exhibit intrinsic robustness, allowing cells to sustain growth despite diverse perturbations. We quantified the inherent robustness of the Saccharomyces cerevisiae genome-scale metabolic network by globally perturbing metabolite production fluxes using a hypothetical sink reaction. Among the 317 high-flux active metabolites, excluding macromolecular intermediates and highly connected cofactors, 85% were found to be robust. Of these robust metabolites, more than half (144/269) were overproduced under perturbation compared with minimal-media controls. These metabolites, mapped to a single central metabolic cluster within the metabolic network, were enriched in core biosynthetic pathways and were largely growth-essential, indicating that the network tolerates elevated biosynthetic demand for most key metabolites. Flux- and pathway-level analyses revealed a coordinated adaptive program involving activation of alternative routes at the network periphery and extensive flux redistribution within central metabolism. Central carbon metabolism and oxidative phosphorylation were broadly suppressed, whereas the pentose phosphate, shikimate, and lipid-related pathways were selectively reinforced to support NADPH generation and redox balance. This reorganization establishes an energy-efficient, redox-stabilized metabolic state that underlies system-wide resilience. Together, these findings show that metabolic robustness emerges from a hierarchical network architecture coupling a stable core with flexible peripheral adaptation. This framework explains cellular resilience and offers design principles for engineering robust, damage-resistant metabolic systems.
|
|
Scooped by
mhryu@live.com
Today, 12:00 AM
|
All eukaryotes, including yeast, plants, animals and humans, possess linear chromosomes. The conserved eukaryotic telomere-telomerase systems, originated and evolved over 1 billion years, protect the chromosomal ends and regulate critical physiological functions through complex networks. In this study, we replace the endogenous eukaryotic telomeres in the single-chromosome yeast Saccharomyces cerevisiae with the prokaryotic telomere system TelN/tos from the Escherichia phage N15, which forms a closed hairpin structure, by interrupting the MRX/Sae2 pathway. The prokaryotic telomeres effectively protect linear chromosomal ends and prevent genetic instability. Through adaptive evolution, we identify yeast strains harboring additional mutations (TEL1 and CYR1) that restore functional MRX/Sae2 activity, thereby improving host fitness and meiotic capacity. Interestingly, the two-associated TelN/tos telomeres position deeper into chromosomes and exhibit increased interactions with their adjacent regions. The successful replacement of a complex eukaryotic chromosomal telomere with a simple bacteriophage system demonstrates functional equivalence between these divergent systems, implying possible natural origins of such stochasticity (e.g., horizontal gene transfers). Furthermore, these engineered strains facilitate development of a tos-YAC system that enabled iterative assembly and stable maintenance of megabase-level heterogeneous DNA (1.23-2.77 Mb), providing a robust platform for large-scale DNA manipulation. Telomeric systems are conserved across eukaryotes and may have originated over 1 billion years ago. Here the authors replaced yeast’s telomeres with a bacterial virus system, resulting in a stable functional assembly of DNA molecules up to 2.77 Mb, offering insights into the possible origins of telomeres.
|
|
|
Scooped by
mhryu@live.com
Today, 5:36 PM
|
The rapid explosion of large-scale, high-throughput biological data has created an urgent demand for efficient analysis pipelines. Traditional bioinformatics approaches, while powerful, often require specialized computational expertise, placing them out of reach for bench biologists. Large Language Models (LLMs) offer new possibilities for automating complex reasoning and tool integration, yet existing LLM-based solutions have not sufficiently lowered this barrier, and expert-level analysis remains inaccessible to most nonexperts. Here, we present BioGAIP, an LLM-powered agent that integrates expert-level reasoning within an end-to-end platform for bioinformatics tasks. By coupling optimized autonomous agents with full graphical interfaces, BioGAIP transforms complex analytical workflows into an automated, user-friendly, and low-intervention process with natural language input. Key features of BioGAIP include dynamic information retrieval, automatic environment configuration, and self-directed design of analysis pipelines, making large-scale multi-omics analysis highly accessible. Built on agent-based client-server architecture, BioGAIP ensures secure resource management and supports heavy computational demands. Extensive evaluations on diverse published datasets demonstrate that BioGAIP reliably recapitulates established biological insights and shows strong potential for novel discovery. By democratizing complex bioinformatics workflows, BioGAIP accelerates accessible data-driven discovery for both experts and nonexperts.
|
|
Scooped by
mhryu@live.com
Today, 2:53 PM
|
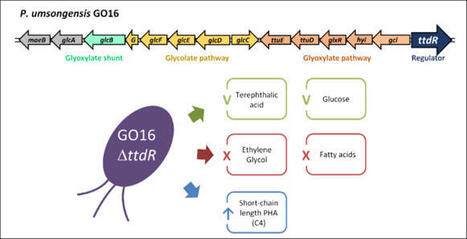
In Pseudomonas umsongensis GO16, a LysR-type transcriptional regulator (LTTRs) ttdR is located 5′ of the gene for glyoxylate carboligase (gcl), involved in ethylene glycol (EG) metabolism. GO16 wildtype (WT) can utilize EG as a sole carbon and energy source but the ttdR knockout strain (ΔttdR) cannot grow on EG, demonstrating its role as an activator in EG metabolism. When ΔttdR was grown with terephthalic acid (TA) or glucose, the growth was comparable to the WT. GO16 originally produces both C4 short-chain-length (scl) PHA and C6-C12 medium-chain-length (mcl) PHA from various carbon sources. Interestingly, ΔttdR produced scl-PHA monomer 5.4-fold and 1.4-fold increased than the WT when TA and glucose were used respectively. Furthermore, ΔttdR exhibited a very long lag phase when grown with fatty acids, namely butyrate or octanoate. This indicates a more complex role of TtdR than simply activating EG metabolism in GO16. The analysis of the GO16 ΔttdR proteome revealed that EG catabolic genes were expressed, but their abundance was 2.8- to 10.5-fold lower (log2) compared to the WT. In addition, higher abundance of enzymes that could be contributing to higher scl-PHA content was also observed. GO16 ΔttdR was further subjected to adaptive laboratory evolution (ALE) with butyrate and octanoate. The evolved strains regained the ability to grow with these fatty acids as the WT, but the mutations accumulated did not confer growth with EG or acetate, suggesting that a mechanism different to glyoxylate shunt has evolved to allow growth with fatty acids.
|
|
Scooped by
mhryu@live.com
Today, 12:41 PM
|
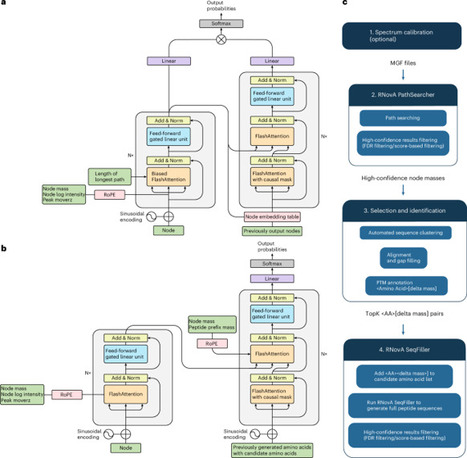
De novo peptide sequencing directly infers sequences from mass spectrometry data without relying on protein databases. Although recent deep learning models can also identify posttranslational modifications (PTMs), they require labeled training data for this task. Here we introduce rotary positional embedding-enhanced de novo sequencing algorithm (RNovA), a transformer-based de novo sequencing algorithm enhanced with relative positional embeddings and a reinforcement-learning-style sequential decision framework. RNovA enables open PTM discovery in a zero-shot setting—without retraining or a predefined list of candidate residues—while maintaining state-of-the-art performance on standard benchmarks. Demonstrating this capability, we successfully identified peptides modified by kynurenine—an uncommon and biologically relevant PTM—in clinical samples from patients with RA and validated this discovery with synthetically synthesized reference peptides. Furthermore, we demonstrated open de novo PTM discovery by analyzing the bacterial strain A1232E, which lacks a reference proteome, and detected an unannotated glutamic acid modification. RNovA enables exploration of previously inaccessible regions of the proteome, including peptides with unexpected or unannotated modifications. RNovA is an open-search de novo peptide sequencing model.
|
|
Scooped by
mhryu@live.com
Today, 11:49 AM
|
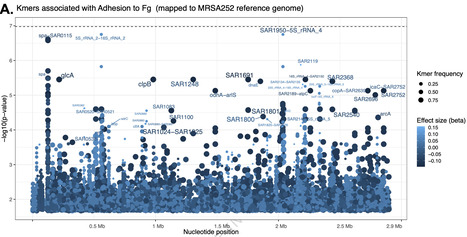
Staphylococcus aureus is a major cause of bloodstream infections, but how variation between strains in their ability to adhere to host proteins influences disease severity remains unclear. Here we show that adhesion is a highly variable and biologically important trait in 236 phylogenetically diverse, representative bacteremia isolates profiled for binding to fibrinogen and fibronectin and analysed together with bacterial whole-genome sequences and matched patients’ clinical data. Stronger fibrinogen-binding, particularly in strains lacking α-toxin, correlates with heightened systemic inflammation (r = 0.401, P = 0.0001) but lower mortality (16.6% vs. 38.7%, P = 0.018), linking bacterial adhesion to distinct clinical outcomes. Genome-wide association analyses identify top-associated variants in genes encoding known adhesins (clfA, fnbA, fnbB, ebh), other surface factors (spa, sdrH), mobile genetic elements, and novel loci (csbB, glcA), although not statistically significant. Functional analyses reveal that protein A limits ClfA-dependent fibrinogen binding through steric hindrance, CsbB interferes with ClfA exposure on bacterial surface, and GlcA enhances fibronectin binding via metabolic regulation. These findings define adhesion as a polygenic, evolutionarily variable trait and suggest that highly-adhesive, α-toxin-defective isolates promote inflammatory but self-limiting infections, whereas weakly adhesive, high-toxicity strains favour immune evasion and severe disease. To uncover how Staphylococcus aureus strain variation influences adhesion and thus disease outcomes, the authors profile 236 bacteremia isolates, showing that high-adhesion, low-toxicity isolates may elicit inflammatory yet self-limiting infections.
|
|
Scooped by
mhryu@live.com
Today, 11:24 AM
|
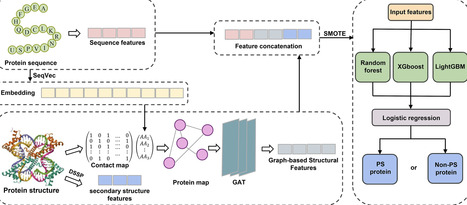
Liquid–liquid phase separation (LLPS) is a key mechanism driving the assembly of membrane-less organelles and is increasingly recognized for its involvement in essential cellular functions and various diseases. However, existing computational approaches largely rely on sequence-level descriptors and often fail to explicitly incorporate structural topology information, limiting their ability to capture the complex determinants of LLPS behavior. Accurate identification of LLPS-capable proteins remains challenging due to their sequence diversity and complex structural determinants. Here, we present MuFGPS (Multi-level Feature Graph-based Predictor for Phase-Separating proteins), a predictive framework integrating sequence-derived physicochemical features, Define Secondary Structure of Proteins-annotated secondary structures, and graph-based structural embeddings from AlphaFold residue contact maps via a multi-head Graph Attention Network. Class imbalance is addressed using Synthetic Minority Oversampling Technique (SMOTE), and classification is performed through a stacking ensemble of Random Forest, XGBoost, and LightGBM. Benchmarks against six representative methods demonstrate that MuFGPS achieves superior performance across all metrics, with notable gains in F1-score and matthews correlation coefficient (MCC). Ablation analyses confirm the synergistic contributions of structural features and ensemble learning to accuracy and robustness. MuFGPS offers a scalable and high-accuracy framework for proteome-wide LLPS protein prediction.
|
|
Scooped by
mhryu@live.com
Today, 11:10 AM
|
Many viral proteins self-assemble into capsid structures, often using their genetic material as a template for assembly. To date, de novo designed capsid-like proteins do not require genetic material as a template for assembly, which can be both an advantage and a disadvantage depending on the use case. Templates are indispensable, for example, in the assembly of linear structures with well-defined lengths. As a first step towards fully de novo designed templated assembly, here we redesign proteins from the Transcription activator-like effector (TALE) family of transcriptional regulators to polymerize on double-stranded DNA (dsDNA) templates. Starting from natural TALE protein sequences, we create idealized repeat proteins with sequence-independent DNA binding properties that self-assemble to form linear protein-DNA complexes with template-controlled lengths. We use high-resolution atomic force microscopy (AFM) and cryo electron microscopy (cryo-EM) to characterize the three-dimensional structures of the DNA-protein hybrid complexes. In these structures, a protein filament helically wraps around the dsDNA similar to natural TALE proteins. As an example application of these materials, we show the system can be used for repetitive peptide antigen display at precisely controlled repeat distances, and that such immunogens elicit robust antigen-specific antibodies in mice. Viral capsids use their genome as an assembly template, but designed proteins lack this feature. Here, the authors redesign TALE proteins to polymerize on DNA, forming linear fibers that display peptide antigens and elicit antibodies in mice.
|
|
Scooped by
mhryu@live.com
Today, 10:40 AM
|
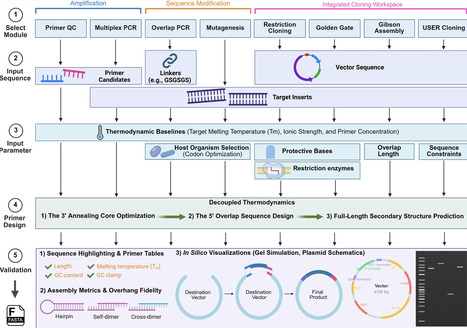
Primer design remains a fundamental yet non-trivial step in modern molecular biology workflows. Although numerous tools are available, they are often fragmented across disparate applications, constrained by commercial licensing, or dependent on external servers, limiting workflow integration and compromising sequence confidentiality. To address this challenge, we developed PrimerWeaver https://ignea.lab.mcgill.ca/primerweaver, a free browser-based tool that integrates primer design, quality control, and support for diverse cloning workflows within a single platform. PrimerWeaver enables overlap PCR, site-directed mutagenesis, multiplex PCR, restriction enzyme-based cloning (including Type II and Golden Gate assembly), and homology-directed methods such as Gibson assembly and Uracil-Specific Excision Reagent (USER) cloning. Primers were generated by optimizing the 3′ annealing region for efficient amplification while appending workflow-specific 5′ sequences according to the selected application. In silico and wet lab validation across diverse cloning workflows demonstrated consistent results relative to established molecular cloning software. All calculations are performed locally in the user’s browser without sequence upload, preserving data confidentiality and supporting reproducible performance across systems. Overall, PrimerWeaver streamlines primer workflows for users ranging from undergraduate students to experienced researchers by integrating multiple PCR and cloning strategies into a single browser-based platform.
|
|
Scooped by
mhryu@live.com
Today, 2:03 AM
|
Genes are connected in complex networks of interactions where often the product of one gene is a transcription factor that alters the expression of another. Many of these networks are based on a few fundamental motifs leading to switches and oscillators of various kinds. And, yet, there is more to the story than knowing which transcription factors control these various circuits. These transcription factors are often themselves under the control of effector molecules that bind them and alter their level of activity. Traditionally, much beautiful work has shown how to think about the stability of the different states achieved by these fundamental regulatory architectures by examining how parameters such as transcription rates, degradation rates, and dissociation constants tune the circuit, giving rise to behavior such as bistability. Such studies, however, explore dynamics without asking how these quantities are altered in real time within living cells as opposed to at the fingertips of the synthetic biologist's pipette or on the computational biologist's computer screen. In this paper, we make a departure from the conventional dynamical systems view of these regulatory motifs by using statistical mechanical models to focus on endogenous signaling knobs such as effector concentrations rather than on the convenient but more experimentally remote knobs such as transcription rates, degradation rates, and dissociation constants. We also contrast the traditional use of Hill functions to describe transcription factor binding with more detailed thermodynamic models. This approach provides insights into how biological parameters are tuned to control the stability of regulatory motifs in living cells, sometimes revealing quite a different picture than is found using Hill functions and tuning circuit parameters by hand.
|
|
Scooped by
mhryu@live.com
Today, 1:55 AM
|
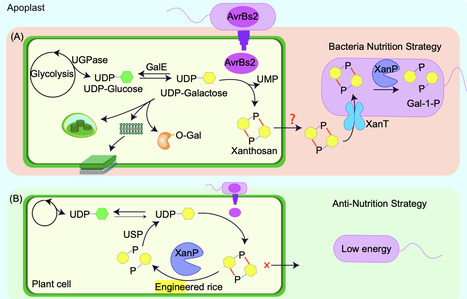
A recent study by Wang et al. found that Xanthomonas exploits host carbohydrates through the conserved effector AvrBs2, which catalyzes UDP-α- D-galactose into xanthosan. Exclusively consumed by the pathogen via XanT and XanP, this ‘nutrition niche’ enables efficient carbon acquisition and virulence, offering new anti-xanthosan strategies for sustainable disease control.
|
|
Scooped by
mhryu@live.com
Today, 1:45 AM
|
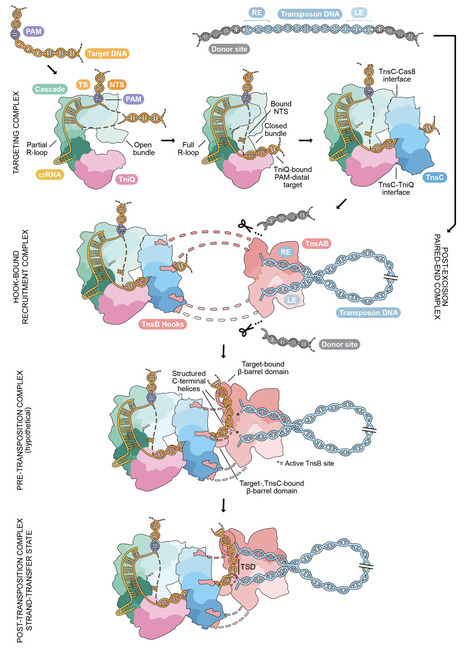
CRISPR-associated transposases (CASTs) achieve site-specific DNA integration by coupling the RNA-guided targeting action of a nuclease-deficient CRISPR-Cas system with the assembly of a Tn7-like transpososome complex. Understanding the detailed mechanisms of this elaborate process is paramount to engineering CAST systems into programmable genetic tools. The type I-F Pseudoalteromonas CAST (PseCAST) displays the highest activity in mammalian cells to date and has been the subject of extensive directed evolution, but efforts to rationally engineer further improvements have been hampered by critical gaps in our understanding of transpososome assembly and activation. Here we use cryo-EM structural analysis, validated by DNA transposition assays, to visualize the PseCAST system in a series of functional states that define the stepwise mechanism of RNA-guided DNA integration. The structure of a target DNA-bound Cascade-TniQ-TnsC complex reveals that conformational changes induced by R-loop formation are coupled to target DNA stabilization and TnsC heptamerization, which in turn recruits the TnsAB transposase via conserved interactions with its C-terminal tail. Finally, the structure of the 1.2 MDa PseCAST transpososome holocomplex reveals specific TnsC-TnsB and TnsB-target DNA interactions that drive allosteric remodelling of the TnsB catalytic site to activate donor DNA integration. Together, these findings establish a unified structural and mechanistic blueprint for RNA-guided DNA integration and lay the foundation for engineering next-generation DNA insertion systems for genome editing applications.
|
|
Scooped by
mhryu@live.com
Today, 1:14 AM
|
Heme is a transformative flavor catalyst for sustainable food systems. This review evaluates recent advances in the metabolic engineering of microbial cell factories, including E. coli, Corynebacterium glutamicum, Bacillus subtilis, Komagataella phaffii, Rhodobacter sphaeroides, and Saccharomyces cerevisiae, for efficient heme biosynthesis. While biosynthetic titers have improved significantly through modular optimization and excretion engineering, heme’s stability and functional mechanisms under industrial food processing conditions remain unsystematically reviewed. In this work, we also emphasize the thermomechanical fate and catalytic role of microbial heme during food processing. We analyze how biochemical pathway diversity (PPD, CPD, and SHD routes) informs host-specific engineering strategies while examining heme’s stability and its role in oxidative cross-linking of proteins under extreme heat and high shear. This review provides a comprehensive framework for engineering next-generation, high-performance sustainable foods.
|
|
Scooped by
mhryu@live.com
Today, 1:06 AM
|
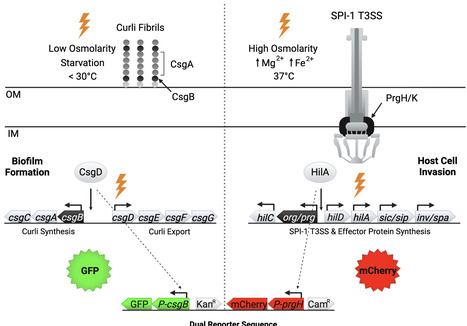
Non-typhoidal Salmonella are a leading cause of foodborne illness. These pathogens cycle between infecting hosts and persisting in the environment as biofilms. Unfortunately, this transition stage is not well understood. In non-typhoidal Salmonella, bistable synthesis of the master biofilm regulator CsgD results in a population split into CsgD-positive biofilm aggregates that synthesize a protective extracellular matrix and CsgD-negative single cells that synthesize the Salmonella pathogenicity island 1 type III secretion system (SPI-1 T3SS), which facilitates host cell invasion. We hypothesize that this is a bet-hedging strategy evolved to improve transmission, providing a way for NTS to cause disease immediately (single cells) or persist over extended periods (biofilms). We built a fluorescent reporter strain of S. Typhimurium to simultaneously track biofilm+ cells (GFP) and SPI-1 T3SS+ cells (mCherry) to better understand the dynamics of bet-hedging. Four S. Typhimurium cell populations were quantified in an in vitro flask model of biofilm development: CsgD+/SPI-1⁻, CsgD⁻/SPI-1+, CsgD+/SPI-1+, and CsgD⁻/SPI-1⁻. We demonstrate that Salmonella population splitting can occur in vivo during infection of C. elegans. At early stages of infection, the worm intestine was mainly dominated by SPI-1 T3SS+ Salmonella cells, while at later time points, both SPI-1 T3SS+ cells and biofilm+ cells are present. The use of the S. Typhimurium dual reporter will allow us to track and better understand the importance of bet-hedging in the Salmonella lifecycle.
|