Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 7:28 PM
|
Random variation in reproductive success--genetic drift--profoundly shapes genetic diversity and evolutionary trajectories. The strength of drift depends on the variance in descendant number, σd2, which governs key evolutionary outcomes: for instance, the establishment probability of a beneficial mutation scales inversely with σd2. However, whether σd2 itself evolves over long timescales has remained unclear, because allele-frequency fluctuations depend on drift only through the effective population size, Ne = N/σd2, which blends census population size with descendant-number variance. Here, we disentangle these components by using model-based Bayesian inference combined with joint tracking of (i) frequency fluctuations of neutrally barcoded lineages and (ii) census population sizes across growth cycles in the E. coli Long-Term Evolution Experiment. Analyzing 33 clones spanning the ancestor through 50,000 generations in two replicate populations (Ara-2 and Ara+2), we find that the strength of genetic drift evolved markedly--and divergently--between the two replicate populations. Both census size and σd2 changed substantially through time, with most variation in Ne driven by shifts in σd2 rather than census size. After approximately 2,000 generations, the σd2 of the two populations diverged sharply: Ara+2 generally remained close to a bottleneck-only null expectation, whereas Ara-2 exhibited 1.5-5x stronger drift, consistent with an evolved increase in stochasticity during growth. Because establishment probability scales as s/σd2, a beneficial mutation of given effect is roughly twice as likely to establish in Ara+2 as in Ara-2. Our results demonstrate that the key parameter governing genetic drift can itself evolve, with direct consequences for adaptation.
|
|
Scooped by
mhryu@live.com
Today, 2:24 PM
|
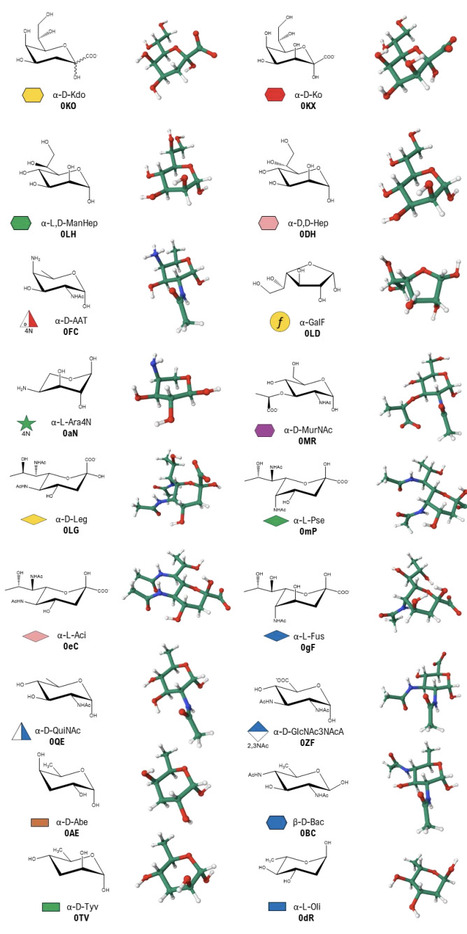
Here we present the GLYCAM Bacterial Carbohydrate Builder, an enhanced version of the GLYCAM-Web Carbohydrate Builder1 structure modeller that integrates support for modelling bacterial glycans, enabling the straightforward generation of three-dimensional structural models. The tool integrates bacterial monosaccharide parametrisations into a curated, user-friendly web-based resource. It provides an intuitive interface for the generation of carbohydrate sequences and generates 3D structural models in PDB file format, as well as the input files required for performing molecular dynamics simulations with the AMBER software package. The current implementation includes a library of 18 bacterial monosaccharides, which can be used in combination with the already parametrised eukaryotic sugars to construct complex bacterial glycans. Common derivatives, including acetylation, methylation, and sulfation are also supported. By validating and integrating bacterial sugar parameters into the GLYCAM-Web Carbohydrate Builder, this work reduces the technical barriers associated with bacterial glycan modelling and facilitates computational studies of complex bacterial glycoconjugates.
|
|
Scooped by
mhryu@live.com
Today, 2:01 PM
|
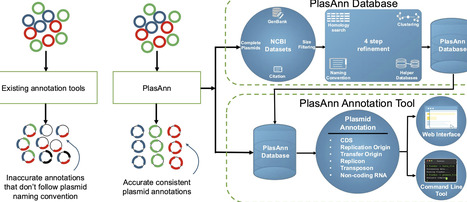
Conjugative plasmids are key drivers of bacterial adaptation, enabling the horizontal transfer of accessory genes within and across diverse microbial populations, yet annotating them remains challenging due to their highly mosaic genetic architectures and inconsistent gene naming conventions that complicate functional predictions and comparative analyses. To address this, we developed PlasAnn, a database designed specifically for genes encoded on natural plasmids, paired with a dedicated annotation pipeline (available via Bioconda or through the URL https://plasann.rochester.edu/). The curated database provides highly accurate, plasmid-type-specific gene names with standardized functional annotations, enabling direct comparison across plasmids without manual curation or specialized expertise, while the integrated annotation tool incorporates other common plasmid features for a fast, one-stop solution that outperforms broad prokaryotic genome annotation pipelines in both accuracy and efficiency. We demonstrate PlasAnn’s utility by showing that plasmid accessory genes from different groups often share conserved repertoires, suggesting dynamic, modular networks of interconnected genes, and by revealing that plasmid-encoded transposable elements frequently carry genes related to bacterial adaptation beyond antibiotic resistance, including metabolism, virulence, and stress responses, emphasizing their broader contributions to fitness and adaptability. These insights, not captured by current field-standard tools, highlight how PlasAnn improves plasmid annotation and advances our understanding of plasmid biology, microbial ecology, and evolution.
|
|
Scooped by
mhryu@live.com
Today, 1:54 PM
|
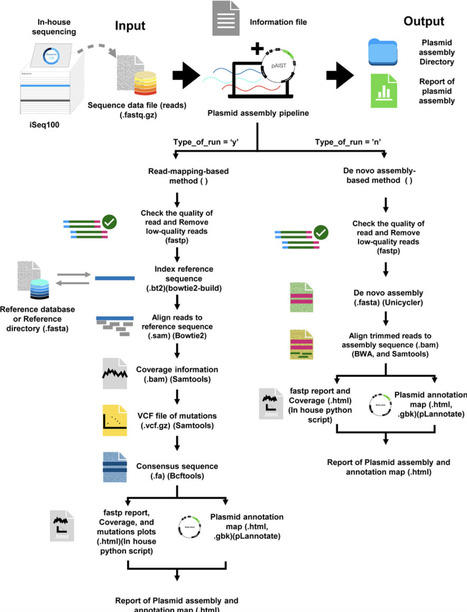
Sequence verification of plasmids is a fundamental process in synthetic biology. For plasmid sequence verification using next-generation sequencing (NGS) library preparation, Tn5 transposase is widely used. Streamlined sequencing workflow for laboratory-scale applications is important; however, recombinant Tn5 production in-house can be laborious. In this study, we demonstrated that the addition of a large soluble tag was not essential for purification and that the fusion of a His10 tag and protein A was sufficient to yield active Tn5 transposase in adequate amounts. In addition, we evaluated exonuclease-based genomic DNA digestion for plasmid sequencing from an E. coli lysate and the data analysis pipeline of sequences derived from the Illumina iSeq100 platform for de novo assembly, reference mapping, and annotation. This study proposes a simple workflow of an in-house easy-to-purify Tn5-based plasmid sequencing platform using a compact benchtop sequencer (AistSeq).
|
|
Scooped by
mhryu@live.com
Today, 12:40 PM
|
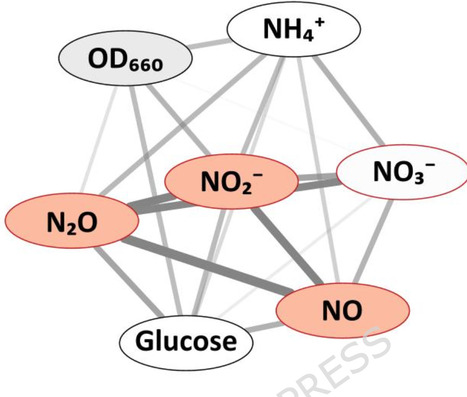
Single-cell protein (SCP) is gaining attention as a source of food and feed, offering a lower environmental footprint than traditional agriculture. Efficient SCP production requires high cell density culturing (HCDC; >20 g cell dry weight L− 1), but O2 supply can become limiting in conventional aerobic systems. To circumvent this bottleneck, we recently proposed an anaerobic strategy using nitrate as an electron acceptor, exploiting denitrification-driven alkalinization in a pH-stat system to regulate substrate provision. Using the model organisms Paracoccus denitrificans and Paracoccus pantotrophus, we achieved biomass concentrations of up to 60 g dry weight L− 1 and protein contents up to 75% (75 ± 5%) in a 3 L fed-batch bioreactor. However, growth rates at high cell density were markedly lower than µmax observed in low-density cultures. In vivo and in silico experiments revealed three interacting constraints: (i) a CO2-pH lag where CO2 accumulation delayed alkalization by denitrification and thus substrate injection; (ii) mixing limitations, leading to poor substrate distribution; and (iii) physiological stress from nitrite accumulating during imbalanced denitrification. The CO2-pH lag emerged as the dominant barrier, resulting in long starvation periods. Lowering the pH setpoint of the pH-stat accelerated CO2 removal and thus substrate provision, but intensified nitrite toxicity. Insufficient mixing compounded the growth limitations as nitrate was only briefly available to a fraction of the population. Small-batch bioassays ruled out accumulation of inhibiting compounds other than nitrite. However, cells grown at high density in the reactor displayed reduced respiration rates, suggesting chronic stress under these conditions. Anaerobic HCDC by denitrification is feasible and yields high-quality biomass, but barriers remain to achieving competitive production rates. The CO2-pH lag appears to be the primary constraint, amplified by incomplete mixing and nitrite toxicity. These factors interact, e.g. mitigating the CO2-pH lag by lowering the process pH exacerbates nitrite toxicity. Future work should integrate reactor engineering to improve mixing and gas removal and strain selection for tolerance to low pH and nitrite, supported by omics and metabolic modelling to understand denitrifier physiology at high cell density.
|
|
Scooped by
mhryu@live.com
Today, 10:44 AM
|
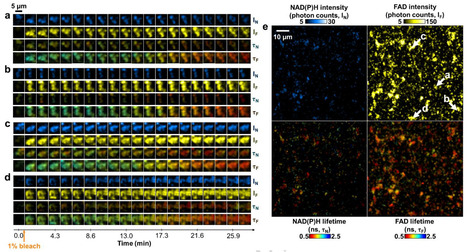
Label-free optical imaging provides non-invasive, high-speed, high-resolution metabolic characterization of live bacteria with single-cell resolution. Here, we demonstrate the ability of label-free multiphoton autofluorescence microscopy to characterize the fast (between 0 and 30 min) metabolic changes in bacteria in response to antibiotic treatments and observe the cell-to-cell metabolic heterogeneity of planktonic bacteria and biofilms. Results indicate that bacteria exhibit a distinct measurable response to bactericidal treatments within seconds. Furthermore, S. aureus biofilms exhibit metabolic heterogeneity, with local pockets of high metabolic activity. Bacteria in biofilms exhibit altered metabolic profiles compared to planktonic bacteria for all four species examined: S. aureus, P. aeruginosa, M. catarrhalis, and S. pneumoniae. These results shed light on the spatial and temporal metabolic heterogeneity of bacteria and the quantification possibilities using label-free nonlinear optical microscopy.
|
|
Scooped by
mhryu@live.com
Today, 10:32 AM
|
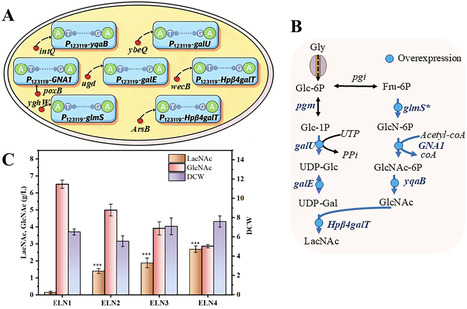
The biosynthetic production of human milk oligosaccharides through metabolic engineering has gained increasing attention in recent years. Yet, limited research has focused on the microbial synthesis of 3′-sialyllactosamine (3′-SLN), a critical intermediate for assembling sialyl Lewis X (sLex). In this work, a pathway enabling 3′-SLN biosynthesis was constructed in E. coli BL21(DE3). A key α2,3sialyltransferase (α2,3SiaT) from Bibersteinia trehalosi was identified and utilized to enhance the sialylation step toward 3′-SLN formation. To improve flux through the N-acetyllactosamine branch, essential genes were chromosomally integrated and overexpressed, while a CMP-N-acetylneuraminic acid synthesis module was coexpressed to secure an adequate donor supply. Furthermore, the expression balance was optimized through combinatorial adjustment of gene copy numbers and translational strength across modules. The engineered E. coli strain produced 0.78 g/L of 3′-SLN in shake-flask cultivation and further accumulated up to 7.75 g/L during 5-L fed-batch fermentation conditions, confirming the feasibility of microbial synthesis of 3′-SLN.
|
|
Scooped by
mhryu@live.com
Today, 10:19 AM
|
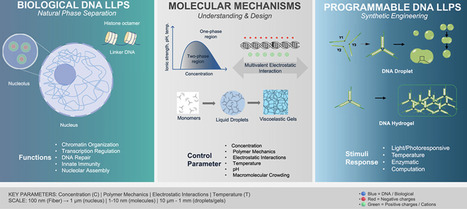
Liquid–liquid phase separation (LLPS) compartmentalizes biological systems into dynamic, membraneless condensates that regulate diverse cellular functions. Although protein and RNA-mediated LLPS have dominated research, DNA increasingly emerges as an active driver of phase separation rather than a passive structural scaffold. DNA-driven condensates orchestrate critical nuclear processes, from chromatin organization and transcriptional regulation to genome stability and innate immune responses. Yet LLPS principles extend beyond biology: programmable DNA nanostructures now enable synthetic droplets and hydrogels with tunable mechanical properties, opening pathways to biomaterials, diagnostics, and synthetic cells. Here we synthesize current understanding of DNA-mediated LLPS across biological and synthetic domains, emphasizing five underappreciated topics: (1) DNA’s active driving role in LLPS through charge and topology; (2) reversible DNA aggregation and aggregate-to-condensate transitions, distinct from irreversible protein misfolding; (3) non-Fickian transport mechanisms including ballistic wave diffusion triggered by molecular recognition; (4) single-molecule mechanical characterization revealing state-dependent material properties; and (5) the multiscale complexity of cellular DNA condensation shaped by topological constraints and hierarchical organization. We highlight emerging single-molecule technologies, optical tweezers, and scanning probe microscopy that directly measure condensate mechanics and state transitions with unprecedented resolution. This integrated perspective bridges fundamental biophysics of natural DNA condensates with rational engineering principles for programmable synthetic systems, providing a blueprint for therapeutic and biotechnological applications.
|
|
Scooped by
mhryu@live.com
Today, 12:38 AM
|
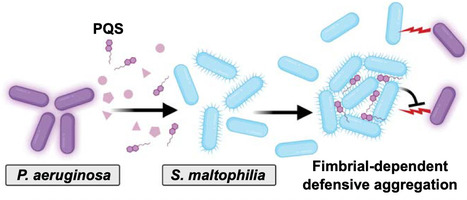
Microorganisms commonly exist in polymicrobial communities, where they can respond to interspecies secreted molecules by altering behaviors and physiology; however, the underlying mechanisms remain underexplored. Here, we investigated interactions between Stenotrophomonas maltophilia and Pseudomonas aeruginosa, coinfecting opportunistic pathogens found in pneumonia and chronic lung infections, such as in cystic fibrosis. We found that S. maltophilia forms large protective multicellular aggregates upon exposure to P. aeruginosa secreted factors. Experimental evolution for lack of aggregation selected for fimbrial mutations and we found that fimbriae are required on both interacting S. maltophilia cells for aggregation. Untargeted metabolomics and targeted validations revealed that the quorum-sensing molecule Pseudomonas quinolone signal (PQS) directly induced S. maltophilia aggregation, and colocalized with the aggregates. Further, in coculture with P. aeruginosa, wild-type S. maltophilia formed aggregates, resulting in up to 75-fold increased survival from P. aeruginosa competition compared to fimbrial mutants. Finally, multiple other bacterial species similarly aggregated upon exposure to P. aeruginosa PQS, indicating a more general response. Collectively, our work identifies a multispecies interaction where a quorum-sensing molecule from a coinfecting pathogen is sensed as a “danger” signal, thereby inducing a protective multicellular response.
|
|
Scooped by
mhryu@live.com
Today, 12:00 AM
|
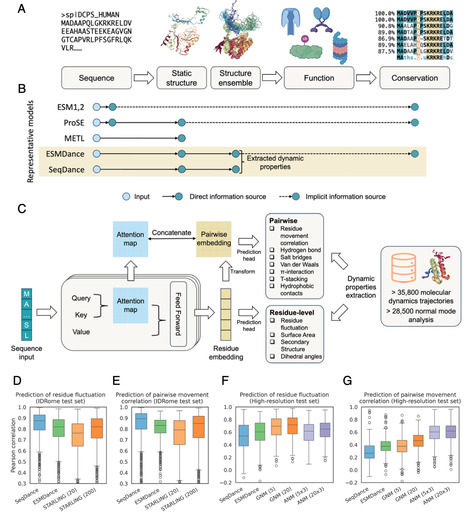
Structural dynamics are fundamental to protein functions and mutation effects. Current protein deep learning models are predominantly trained on sequence and/or static structure data, which often fail to capture the dynamic nature of proteins. To address this, we introduce SeqDance and ESMDance, two protein language models trained on dynamic biophysical properties derived from molecular dynamics simulations and normal mode analyses of over 64,000 proteins. Both models can be directly applied to predict dynamic properties of unseen ordered and disordered proteins. SeqDance, trained from scratch, has attentions that capture dynamic interaction and comovement between residues, and its embeddings encode rich representations of protein dynamics that can be further utilized to predict conformational properties beyond the training tasks via transfer learning. SeqDance predicted dynamic property changes reflect mutation effect on protein folding stability. ESMDance, built upon ESM2 (Evolutionary Scale Model II) outputs, substantially outperforms ESM2 in zero-shot prediction of mutation effects for designed and viral proteins which lack evolutionary information. Together, SeqDance and ESMDance offer a framework for integrating protein dynamics into language models, enabling more generalizable predictions of protein behavior and mutation effects.
|
|
Scooped by
mhryu@live.com
January 26, 11:36 PM
|
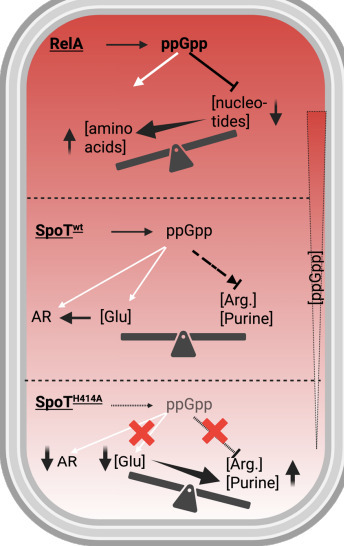
Bacteria survive fluctuating and hostile environments by coordinating core metabolism with stress-protective responses. How these processes are integrated at the molecular level remains incompletely understood. Here, we uncover a previously unrecognized role for the stringent response enzyme SpoT in coordinating amino acid homeostasis and acid resistance through maintenance of a basal pool of the alarmone ppGpp. Focusing on a conserved histidine residue in SpoT (H414), we show that its disruption causes severe growth defects in minimal medium and catastrophic acid sensitivity in E. coli, independently of the canonical RelA-mediated stringent response. Transcriptomic and genetic analyses reveal that a SpoT H414A substitution leads to misallocation of metabolic flux, characterized by excessive arginine biosynthesis and concomitant depletion of intracellular glutamate, the essential substrate for the glutamate-dependent Gad acid resistance system. Intragenic suppressor mutations within spoT restore growth and acid resistance by re-establishing a sub-basal yet physiologically sufficient ppGpp pool, demonstrating that very low levels of ppGpp are required for metabolic balance and stress preparedness. Importantly, the requirement for SpoT H414 in supporting growth and acid survival is conserved in pathogenic Salmonella and Shigella. Together, our findings establish SpoT-derived basal ppGpp as a distinct regulatory regime that integrates metabolism and acid resistance, providing a framework for understanding how bacteria maintain physiological homeostasis and survive acidic environments.
|
|
Scooped by
mhryu@live.com
January 26, 11:27 PM
|
Multiple sequence alignment (MSA) data underlies current principles in protein folding and protein-protein interaction prediction, from which large language models (LLMs) in tandem with protein datasets, can predict protein structure. However, what is missing are user-friendly tools that enable researchers to predict and demonstrate coevolution - the principal input which these MSAs infer. Here we present tools to identify and visualize coevolution, through a pipeline (CoEVFold) that uses basic direct coupling algorithms derived from GREMLIN and alignment of sequences from MMSEQs2. The pipeline generates a visual representation of coevolution for a single protein but can also represent coevolution of homomeric or heteromeric protein complexes, as well as coevolution within protein networks. The input for this pipeline can be an amino acid sequence, or user input protein structures from Alphafold their own files or the PDB database. In validation of CoEVFolds capabilities, and utilizing proteins from known prokaryotic and eukaryotic model systems (Bacillus subtilis, Escherichia coli and Saccharomyces cerevisiae), as well as phage proteins, CoEVFold predicts coevolution between proteins known to interact, proteins known to oligomerize, and coevolution in proteins known to be part of a protein complex. Collectively, these suite of tools, named CoEVFold suite, have broad applicability and provide a useful toolkit to those interested in dissecting protein-protein interactions and networks. https://colab.research.google.com/drive/1MSSvNTq7KZ4Lr0XTz89vUuK-J3xOTzwS?usp=sharing and Github.
|
|
Scooped by
mhryu@live.com
January 26, 11:22 PM
|
Identifying common function-determining structural motifs among proteins with different folds is a foundational task in computational biology with no go-to solution. Indeed, standard alignment tools like TM-align are ill-suited for matching small, sequence-order-independent motifs, while specialized tools have limited success. Here, we introduce LocAlign, a local structural alignment algorithm based on geometric deep learning. Given two protein structures, LocAlign iteratively predicts atom-level correspondences and a 3D superimposition. By formulating training as a weakly supervised task on pairs of proteins bound to identical ligands, we bypass the need for ground-truth alignments. We find that LocAlign recovers known functional motifs without explicit supervision, identifying high-quality alignments for 87% and 37% of protein pairs with similar and dissimilar folds, respectively. We show that, equipped with confidence scoring and motif-conditioning capabilities, LocAlign supports diverse applications, including functional annotation of the dark proteome and drug off-target screening. LocAlign is thus a potent, versatile framework for protein functional site comparison.
|
|
|
Scooped by
mhryu@live.com
Today, 7:22 PM
|
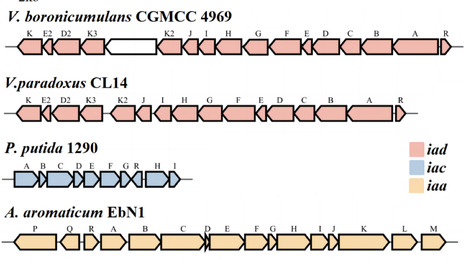
Plant growth-promoting rhizobacteria (PGPR) of the genus Variovorax facilitate plant growth through beneficial microbe-plant interplay. Unlike most PGPR, Variovorax boronicumulans CGMCC 4969 utilizes indole-3-acetonitrile as a precursor to generate indole-3-acetic acid (IAA), which is subsequently metabolized by itself. In this study, IAA enhanced the growth of V. boronicumulans CGMCC 4969 in minimal salt medium (MSM), whereas it inhibited bacterial growth when glucose was added to the MSM broth. IAA was rapidly degraded within 12 h in MSM broth despite glucose appeared or not. Notably, in LB broth, the cell growth was significantly inhibited by IAA concentration beyond 1 mmol/L, while the IAA degradation capability of CGMCC 4969 was significantly increased following exposure to IAA-dosed LB medium. V. boronicumulans CGMCC 4969 degraded IAA to yield a new intermediate 3-hydroxy-anthranilate. An iad gene cluster was identified in V. boronicumulans CGMCC 4969, and co-expression of the iadD and iadE genes endows E. coli with the capacity to degrade IAA. This degradation efficiency is augmented when the iadC gene is expressed simultaneously. Subsequent proteomics and bioinformatics analyses highlighted that the addition of IAA induced a significant up-regulation of ABC transporter proteins, in particular IadK3 and IadK2. Interestingly, there was also a significant increase in protein expression associated with group-sensing metabolism. Collectively, this research helps our understanding of the intricate regulatory mechanisms of IAA within Variovorax own metabolism and expands our knowledge of its complex role in plant-microbe interactions. Key points The iad gene cluster degraded IAA to a previously uncharacterized intermediate.Adding IAA during the cell culture period enhances IAA-degrading activity.Proteomics defined the adaptive response to IAA.
|
|
Scooped by
mhryu@live.com
Today, 2:18 PM
|
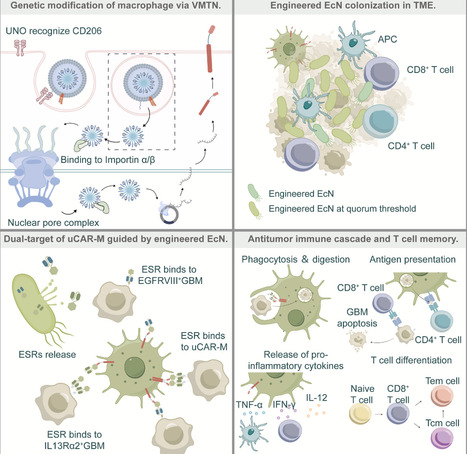
Glioblastoma (GBM) remains a highly lethal form of cancer due to its molecular heterogeneity and the immunosuppressive microenvironment surrounding the tumor. Here, we report a modular immunotherapy platform characterized by its flexibility to simultaneously target multiple antigens. Specifically, we utilize engineered E. coli Nissle to colonize tumors and produce bispecific engagers that simultaneously target EGFRvIII and interleukin (IL)-13Rα2. These tags direct in situ-reprogrammed chimeric antigen receptor (CAR) macrophages, which are edited using nanoparticles and delivered within a shear-thinning hydrogel, to execute targeted phagocytosis. This probiotic-macrophage crosstalk eliminates tumor cells while converting protumor M2 macrophages into immunostimulatory M1 effectors. In aggressive orthotopic GBM mouse models, this strategy achieves 83% survival at the 120-day endpoint, representing a 5-fold improvement over single-target controls and establishing durable immunological memory that effectively combats recurrence. By functioning as multifunctional immune hubs, this platform offers a versatile framework designed to overcome the antigenic complexity of solid tumors.
|
|
Scooped by
mhryu@live.com
Today, 1:59 PM
|
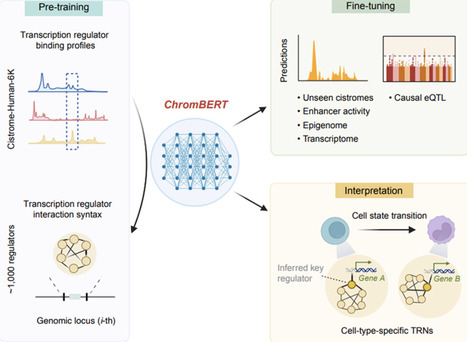
Gene expression is shaped by transcriptional regulatory networks (TRNs), where transcription regulators interact within regulatory elements in a context-specific manner. Deciphering context-specific TRNs has long been constrained by the severe sparsity of cell-type-specific chromatin immunoprecipitation sequencing (ChIP-seq) profiles. Here, we present ChromBERT, a foundation model pre-trained on large-scale human ChIP-seq datasets covering ∼1,000 transcription regulators. ChromBERT learns the genome-wide syntax of regulatory cooperation and generates interpretable TRN representations. After prompt-enhanced fine-tuning, it outperforms existing methods for imputing unseen cistromes. Moreover, lightweight fine-tuning on cell-type-specific downstream tasks adapts the TRN representations to capture regulatory effects and dynamics within any given cellular context. The resulting context-specific representations can then be interpreted to infer regulatory roles of transcription regulators underlying these cell-type-specific regulatory outcomes without requiring additional ChIP-seq experiments. By overcoming the limitations of sparse transcription regulator data, ChromBERT significantly enhances our ability to model and interpret transcriptional regulation across a wide range of biological contexts.
|
|
Scooped by
mhryu@live.com
Today, 1:00 PM
|
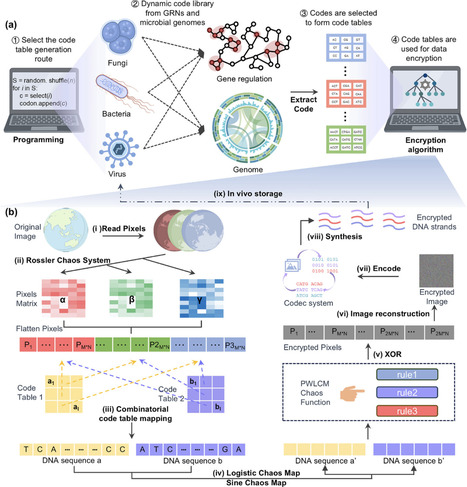
DNA is a promising medium for next-generation data storage because of ultrahigh information density and stability. DNA storage within living organisms presents further advantages, such as self-replication, compactness, and concealment. Early efforts primarily developed predetermined methods for encoding and decoding data using in vivo DNA sequences. However, these methods may pose a security risk while opening a clear channel for potential data access and breaches. To address these challenges, we propose a unified paradigm, integrated computational–biological programming (ICBP), by exploiting the intrinsic digital characteristics within computational and microbial systems. ICBP involves the construction of dynamic code tables from gene regulatory networks or complete genomes across diverse species, expanding the key space by more than 100 orders of magnitude compared with existing methods. The encryption algorithm in ICBP benefits from DNA encoding, computing, and computational operations, leading to superior encryption quality and resistance to brute force and statistical attacks. Furthermore, we demonstrated the practical utility of ICBP via the successful encryption, microbial storage, and decryption of digital files within living systems, achieving 100% data recovery after 100 generations of replication. By combining computational logic with the biological complexities of living systems, the ICBP offers a transformative strategy for secure DNA data storage.
|
|
Scooped by
mhryu@live.com
Today, 10:45 AM
|
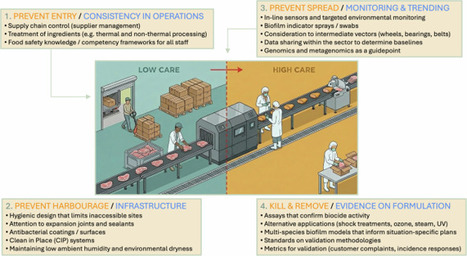
Food safety risks are controlled in agrifood settings by reducing the microbial burden in food ingredients and food production environments. Hygiene programmes involve an incremental implementation of chemical treatments (e.g., disinfectants) and engineering controls (e.g., elimination of susceptible harbourage sites). The strategies to disrupt the presence and transmission of microbial risks to foods are being refined by advanced microbiology and genomics that provide actionable evidence on the precise nature of local ecologies.
|
|
Scooped by
mhryu@live.com
Today, 10:36 AM
|
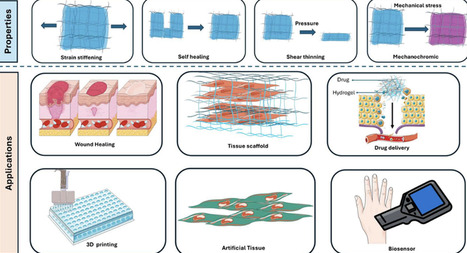
Mechanoresponsive biomaterials are a revolutionary class of materials designed to respond dynamically to mechanical stimuli, providing tissue engineering and regenerative medicine with precise control over biological processes. Through processes including supramolecular interactions, strain stiffening, and force-induced conformational changes, these materials, which include hydrogels, elastomers, and piezoelectric composites, imitate the mechanics exclusive to biological tissues. Additionally, mechanoresponsive systems improve drug delivery by releasing drugs in response to pH changes or mechanical strain using various materials, including magnetic scaffolds and ultrasound-triggered micelles. Despite advancements in numerous arenas of biological sciences, problems with clinical translation, scalability, and long-term biocompatibility still exist. New developments combine technologies like 4D bioprinting to create dynamic, patient-specific scaffolds and artificial intelligence (AI)-assisted design to maximize material qualities. To achieve material innovation with the desired level of biological complexity, future initiatives should focus on multifunctional platforms that combine mechanical, electrical, and biochemical inputs at an advanced level. This review dives into several aspects of mechanoresponsive biomaterials by navigating through the fabrication methods, underlying principles, inception of these in biomedical applications, and progression through the current research settings.
|
|
Scooped by
mhryu@live.com
Today, 10:24 AM
|
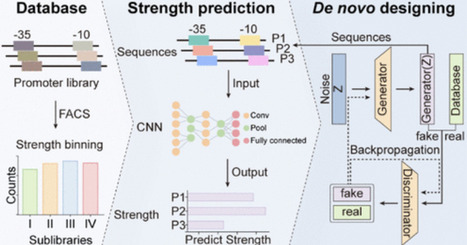
Promoters are essential in transcriptional regulation, with the −10 and −35 boxes playing a critical role in determining their strength. Modulating these regions can effectively fine-tune promoter strength. However, the lack of a clear quantitative relationship between sequence composition and transcriptional output impedes the rational design of promoters. To address this, we developed a synthetic promoter library by varying RNA polymerase binding energies at the −10 and −35 boxes. The library was partitioned into four sublibraries with expression strengths spanning an 80-fold range. Using fluorescence-activated cell sorting FACS followed by sequencing, we identified 20,799 distinct promoters. Analysis of this library uncovered distinct sequence-activity patterns, including a small subset of −35 box sequences that consistently conferred high transcriptional output across diverse −10 partners. Based on this, we developed an artificial intelligence platform that integrates a convolutional neural network for strength prediction (Pearson’s r = 0.84) with a balanced generative adversarial network incorporating a gradient penalty for de novo promoter design. By coupling these models, we achieved a precise design of promoters with user-defined strengths (r = 0.85), establishing a bidirectional framework that links −10/–35 boxes to transcriptional activity through deep learning. This study expands the sequence diversity of functional −10 and −35 boxes in E. coli, provides a predictive platform for rational promoter engineering, and deciphers combinatorial motif interactions governing transcriptional regulation.
|
|
Scooped by
mhryu@live.com
Today, 12:43 AM
|
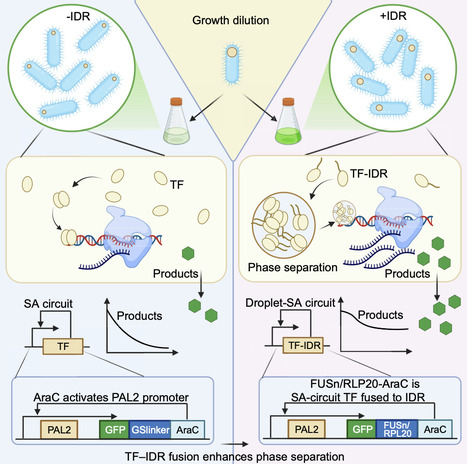
The team engineered transcription factors (TFs) by co-opting biomolecular phase separation to form liquid-like condensates that function as intracellular reservoirs, locally concentrating the transcriptional machinery during cell proliferation. This elegant method focuses on fusing TFs to intrinsically disordered regions (IDRs), particularly, the well-characterised N-terminal domain of FUS protein (FUSn) or synthetic resilin-like polypeptides (RLP20). These IDRs mediate weak, multivalent interactions that drive the de novo formation of dynamic, membraneless organelles within the bacterial cytoplasm. The resulting transcriptional condensates enrich TFs at the promoter regions. Thus, they effectively decouple the local transcriptional activity from the declining average cellular TF concentration, thereby preserving gene expression programs and complex circuit functionalities, such as memory.
|
|
Scooped by
mhryu@live.com
Today, 12:33 AM
|
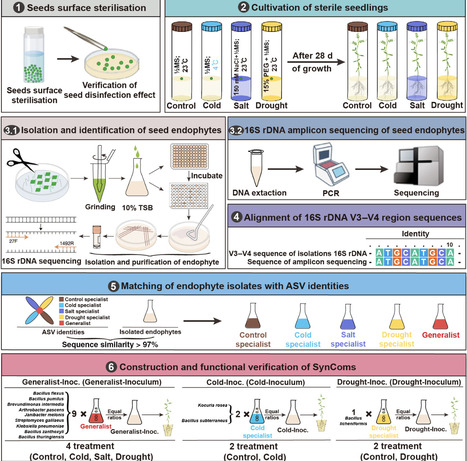
The plant and its associated microbiota constitute a holobiont. Within this framework, the seed endophyte reservoir, shaped through multigenerational selection, exhibits pronounced host specificity, mutualistic potential, and signatures of co-evolution. We hypothesise that this reservoir operates as a ‘symbiotic toolbox’ forming an ‘Anticipated Utility Microbiota’ within the holobiont. Upon germination, specific microbes from this toolbox may undergo resuscitation to buffer environmental stresses, thereby influencing plant fitness. Using axenic Vicia sativa seeds, we simulated cold, salinity, and drought stresses and applied 16S rRNA sequencing to track seed symbiont resuscitation. Taxa showing resuscitation across stresses were classified as generalists, whilst those resuscitating under specific stresses were specialists. Microbial inoculants from these taxa were then tested in pots for host growth effects. As expected, distinct resuscitation patterns under different stresses supported the hypothesised seed ‘symbiotic toolbox’. We identified 115 generalist amplicon sequence variants (Methylobacterium, Pantoea, and Sphingomonas) and stress-specific specialists: 60 cold specialists (Stenotrophomonas and Geobacter), 79 salt specialists (Leptotrichia), and 13 drought specialists (Proteobacteria). Strikingly, generalist microbial inoculants consistently promoted seedling growth across stresses, whilst specialist inoculants showed stress-specific efficacy. This study elucidates a holobiont mechanism whereby vertically transmitted seed microbes constitute a ‘symbiotic toolbox’ that differentially resuscitates under stress, thereby enhancing seedling fitness.
|
|
Scooped by
mhryu@live.com
January 26, 11:52 PM
|
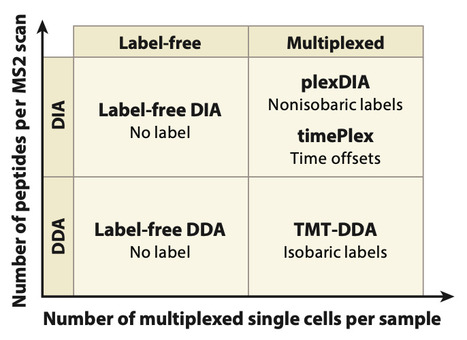
Over the last decade, proteomics analysis of single cells by mass spectrometry transitioned from an uncertain possibility to a set of robust and rapidly advancing technologies supporting the accurate quantification of thousands of proteins. We review the major drivers of this progress, from establishing feasibility to powerful and increasingly scalable methods. We focus on the trade-offs and synergies of different technological solutions within a coherent conceptual framework, which projects considerable room both for throughput scaling and for extending the analysis scope to functional protein measurements. We highlight the potential of these technologies to support the development of mechanistic biophysical models and to help uncover new principles.
|
|
Scooped by
mhryu@live.com
January 26, 11:30 PM
|
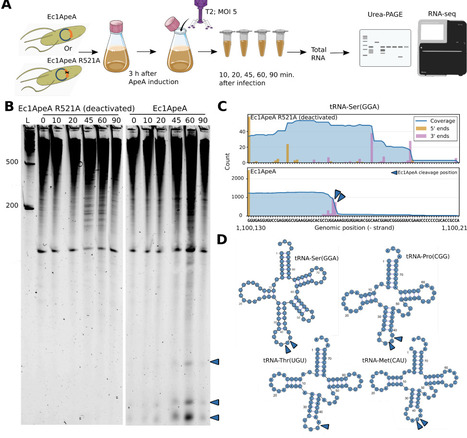
Bacteria and archaea encode diverse antiviral defense systems, many of which rely on toxic effector proteins that are activated specifically upon bacteriophage infection. However, the mechanisms by which infection is recognized and coupled to effector activation remain poorly understood for most antiviral systems. Here, we focus on ApeA, a HEPN-domain antiphage protein that confers immunity through cleavage of host tRNAs within their anticodon loops. We show that ApeA proteins form large doughnut-shaped oligomers that are activated upon ligand binding in a conserved protein pocket distinct from the catalytic center. In the Ec2ApeA variant, this pocket specifically recognizes 5′-phosphorylated deoxydinucleotides that likely arise as intermediates of host genome degradation by viral nucleases, thereby enabling Ec2ApeA to achieve a broad protection profile. Together, our results reveal how small-molecule products of virus-induced host cell destruction function as signals that activate bacterial immune defenses.
|
|
Scooped by
mhryu@live.com
January 26, 11:24 PM
|
Iron acquisition is critical to bacterial growth and pathogenesis. Here, we describe a previously unrecognized mechanism by which the major human pathogen Pseudomonas aeruginosa acquires iron through a unique partnership with bacteriophages (phages). Pf is a filamentous phage that infects P. aeruginosa and is associated with chronic infections. We reveal that Pf contributes to P. aeruginosa pathogenesis by promoting iron uptake. We demonstrate that Pf phage is highly induced under iron-deplete growth conditions and that once induced, Pf virions directly bind and locally concentrate free iron. Pf-bound iron is more efficiently utilized by P. aeruginosa than unbound iron, enhancing bacterial growth. We further demonstrate that Pf-mediated iron acquisition depends on type IV pili, which facilitate Pf attachment and confers strain selective uptake of phage-bound iron, providing a competitive fitness advantage in polymicrobial settings. Together, these findings identify an unrecognized role for filamentous phages in mediating iron acquisition and reveal a novel phage bacterium partnership that operates through selective kin cooperation.
|