 Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 10:54 AM
|
Integrases serve as powerful biotechnology tools that catalyze recombination at specific DNA sequences (att sites) and facilitate chromosomal integration of gene cargos transferred into cells. Given that genomes often lack the attB integration sites recognized by frequently utilized integrases, integrase technology has largely been restricted to genetic engineering of model organisms into which attB sites can be synthetically introduced. To enable single-step site-specific integrase-mediated genome editing in a broad spectrum of prokaryotes, we have devised the Integrase-On-Demand (IOD) method. IOD systematically identifies integrases, within bacteria and archaea, that can integrate into available attB sites in any target prokaryote. Computational results show that diverse bacteria generally have multiple potentially useable native attB sites for novel integrases. We confirmed the functionality of predicted integrase and attB pairs for mediating site-specific integration of heterologous DNA into the genomes of Pseudomonas putida S12 and KT2440 and Synechococcus elongatus UTEX 2973, measuring efficiency of integration using nonreplicating vectors. By eliminating the requirement to introduce non-native attB sites into the target genome, IOD may, when suitable transformation methods exist, allow facile genome integration of large constructs in nonmodel and possibly nonculturable bacteria.
|
|
Scooped by
mhryu@live.com
Today, 10:22 AM
|
Rice paddies are a major, persistent source of atmospheric methane (CH4), emission rates depend on the partitioning of photosynthate carbon between the rice plant and the rhizosphere microbiome. Although ratoon season rice (RR) is shown to emit far less CH4 than main-crop rice (MC), the mechanisms have remained unresolved. This work conducts a 2-year field experiment in which RR is compared with MC and with late rice (LR) synchronized to the RR heading stage. Relative to MC and LR, RR lowers daily CH4 flux by 91%, raises daily grain yield by 34%–57%, and increases net economic return by 90%–136%. Mechanistically, 13C-labelling reveals that RR diverted more newly fixed carbon to the grain and less to the rhizosphere, thereby restricting acetate availability for methanogens. Rhizosphere metagenomics show reduced abundance of Methanobacteriaceae and down-regulation of methanogenic genes in RR. This carbon-reallocation pattern is underpinned by an abscisic acid (ABA)-mediated interaction between OsCIPK2 and OsSWEET1A, which simultaneously curtailed carbon efflux from roots and enhanced grain filling. This study is the first to establish a comprehensive framework of “ABA regulation—carbon allocation—microbial function—emission reduction and efficiency enhancement.” It provides targetable strategies for carbon allocation and microbial management within climate-smart rice farming systems.
|
|
Scooped by
mhryu@live.com
Today, 10:08 AM
|
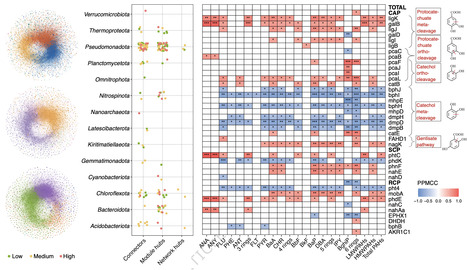
Polycyclic aromatic hydrocarbons PAH are persistent coastal pollutants, yet the ecological and genomic strategies governing the persistence and function of degraders under in-situ stress remain elusive. Here we decode these adaptive mechanisms using metagenomics and cultivation in the Pearl River Estuary, integrated with global comparative analyses. We reveal a multi-level adaptive strategy where high PAH stress drives ecological network densification and selectively enriches the thermodynamically favorable catechol ortho-cleavage pathway. Crucially, we elucidate a conserved genomic “division of labor”, where chromosomes encode stable upstream activation steps, while plasmids serve as specialized, mobile reservoirs for downstream central aromatic processing. This plasmid-mediated functional partitioning is globally conserved across diverse coastal ecosystems, although the reliance on mobile vectors is dynamically tuned by environmental stability. Collectively, these findings unveil a holistic adaptive framework that integrates ecological cooperation with genomic partitioning, highlighting a plasmid-mediated “plug-and-play” mechanism that underpins microbial resilience and guides precision bioremediation. Coastal microbiomes adapt to polycyclic aromatic hydrocarbon stress via a conserved genomic division of labor where chromosomes retain upstream specificity, while plasmids mobilize downstream detoxification modules, shown by sampling in China’s Pearl River Estuary.
|
|
Scooped by
mhryu@live.com
February 8, 10:11 AM
|
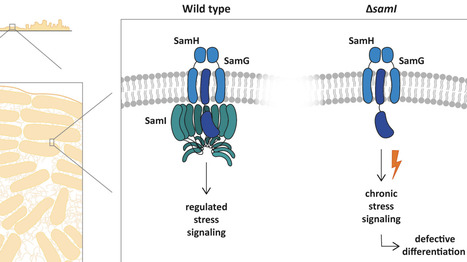
Bacillus subtilis adapts to fluctuating environmental stress, such as membrane perturbation or alkaline conditions, using membrane associated regulatory complexes. Here, we rename the previously termed pspA ydjGHI operon to pspA samGHI (for starvation and motility) to reflect its functional roles in membrane envelope stress signalling. The SamG SamH membrane proteins recruit SamI, a cytosolic SPFH protein, which stabilizes focal membrane localization and recruitment of PspA, an ESCRT III homolog. Under normal conditions, this system transiently assembles at the membrane, stabilizing it and allowing proper motility, secretion, and biofilm formation. Loss of SamI (ΔsamI/ΔydjI) leads to unbalanced SamG SamH activity leading to a constitutive stress signalling, and global transcriptional changes reminiscent of starvation situations. This, in turn, blocks secretion of the matrix protein BslA, preventing biofilm formation, and reducing motility. Deletion of samH in combination with ΔsamI restores biofilm formation, while ΔpspA mutants form biofilms normally, indicating that PspA is dispensable for the developmental phenotype. Our findings reveal that beside membrane integrity SamGHI coordinates transcriptional homeostasis and multicellular development through formation of a membrane integral stress sensor complex.
|
|
Scooped by
mhryu@live.com
February 8, 9:53 AM
|
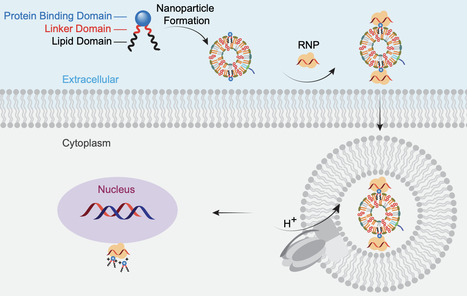
CRISPR/Cas9-based gene-editing technologies offer promise for treating inherited retinal diseases (IRDs), however safe and efficient ocular delivery of precision editors remains challenging. To address this challenge, we report a class of Coomassie brilliant blue (CBB)-derived lipidoids that bind and deliver proteins. Subretinal injection of Cre complexed with these lipidoids into mT/mG mice leads to robust recombination in the retinal pigment epithelium and photoreceptors. We employ the CBB-lipidoid platform to deliver adenine base editor (ABE) ribonucleoproteins (RNP). Incorporating CBB lipidoids into liposomes improves delivery efficiency. CBB11 stands out for facilitating precise in vivo ABE-mediated gene editing. Delivery of liposome-CBB11-RNP complexes results in a 120-fold increase in base editing compared to RNP alone and restores the scotopic ERG b-wave response in the rd12 mouse model. These results demonstrate the potential of CBB-augmented, liposome-RNP systems for therapeutic gene editing in the eye, paving the way for single-dose precision medicines to treat IRDs. Precise and efficient CRISPR genome editing requires specialized delivery systems. Here, the authors develop Coomassie lipidoids that deliver purified adenine base editors into retinal tissues, making it possible to achieve robust genome editing with a defined, non-viral nanomedicine.
|
|
Scooped by
mhryu@live.com
February 8, 12:40 AM
|
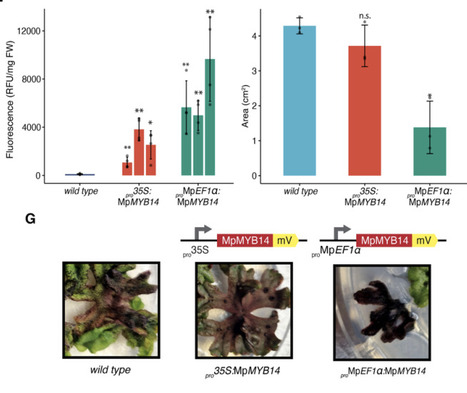
Plant-derived pigments offer sustainable alternatives to synthetic colorants, yet their practical deployment in textiles is limited by restricted chemical diversity and low abundance. Liverworts represent a source of diverse chemical compounds, and the model liverworts Marchantia polymorpha is an emerging as chassis for bioengineering and synthetic biology. Here, we report the biotechnological application of auronidins, a rare class of flavonoid pigments, as textile dyes. Using the Marchantia, we engineered enhanced auronidin production through controlled expression of the R2R3-MYB transcription factor MpMYB14. We systematically benchmarked constitutive and inducible gene expression systems, including heat-shock, glucocorticoid receptor, and β-estradiol (XVE) circuits, identifying inducible strategies that decouple biomass accumulation from secondary metabolite production while achieving high pigment yields. Extracted auronidins were used to dye cotton yarn directly, demonstrating the feasibility of auronidins for textile dyeing. Our results establish Marchantia as a versatile plant chassis for programmable secondary metabolite production and introduce auronidins as a promising natural pigment platform for sustainable textile biotechnology.
|
|
Scooped by
mhryu@live.com
February 8, 12:34 AM
|
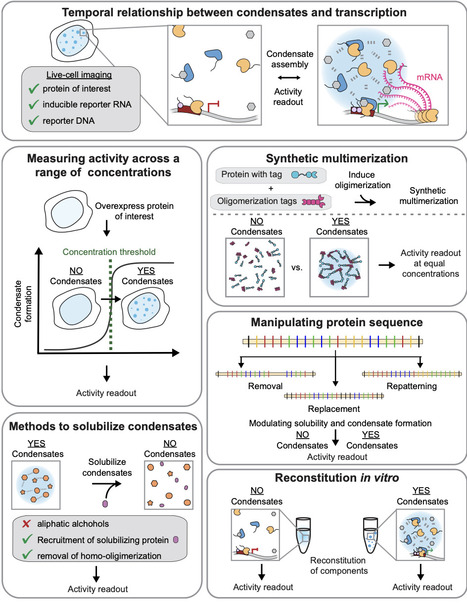
Transcription of the right genes at the right time is crucial for physiology and often dysregulated in disease. For a process that requires precise outcomes, the underlying molecular interactions regulating transcription can be surprisingly dynamic and short-lived. Various sources of multivalency have been proposed to stabilize short-lived interactions at specific genomic loci. Through multivalent contacts, components of the transcription machinery form complexes and, above-threshold concentrations, can form condensates by undergoing reversible density transitions (i.e., phase separation). Importantly, coupling the density transition to networking generates a condensate-spanning network with emergent properties that soluble complexes do not possess, including enhanced dwell times of relevant components, distinct chemical environments, capillary forces, and biochemically active interfaces. Here, we will review the current evidence for and against the role of density transitions in regulating transcription, discuss best practices for studying the functional roles of these phenomena, and consider how emergent properties may regulate transcription.
|
|
Scooped by
mhryu@live.com
February 8, 12:20 AM
|
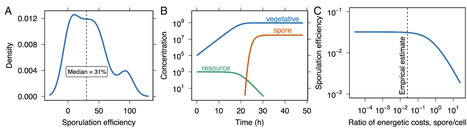
Energy is required for the expression and maintenance of complex traits. In many habitats, however, free energy available to support biosynthesis is in vanishingly short supply. As a result, many taxa have evolved persistence strategies that support survival in unfavorable environments. Among these is sporulation, an ancient bacterial program governed by a large genetic network that requires energy for both regulation and execution. Yet sporulation is a last resort, initiated when cellular energy is nearly exhausted. To resolve this paradox, we quantified the energetic cost of sporulation in units of ATP by integrating time-resolved genome, transcriptome, and proteome profiles. The full cost of the spore cycle, including both formation and revival, ranks among the most energy-intensive processes in the bacterial cell, requiring almost 1010 ATP and consuming about 10% of the total energy budget. The majority of this cost arises from translation, membrane synthesis, and protein turnover. Despite its considerable upfront investment, sporulation enables long-term survival and becomes optimal when harsh conditions extend over timescales of months or longer. This trade-off between immediate cost and delayed benefit helps explain when sporulation is maintained or replaced by alternative strategies. By incorporating our estimates into mechanistic models, we show how metabolic constraints shape sporulation efficiency, while genome-wide mutation accumulation data reveal that even modest energetic burdens can become visible to selection, influencing the evolutionary fate of this complex and widespread trait.
|
|
Scooped by
mhryu@live.com
February 8, 12:06 AM
|
Traditional metabolic engineering faces numerous challenges in constructing microbial cell factories, such as inefficient gene editing, time-consuming and labor-intensive screening processes, and the complexity of multi-gene optimization. High-throughput (HTP) genome editing technology accelerates the optimization of microbial metabolic pathways by precisely and efficiently modifying multiple genes. Furthermore, HTP genome editing technology enables the rapid screening and modification of key enzymes or regulatory factors across multiple metabolic pathways, facilitating the analysis of complex regulatory mechanisms. These advantages make it a key enabling tool for both top-down analysis and bottom-up assembly of metabolic pathways. This review summarizes the mechanisms and applications of various HTP genome editing and discusses the development of HTP strategies that accelerate the design-build phase for microbial cell factories (MCFs). These advancements promised to significantly enhance the performance of MCFs and drive the next generation of sustainable, bio-based production technologies.
|
|
Scooped by
mhryu@live.com
February 7, 3:17 PM
|
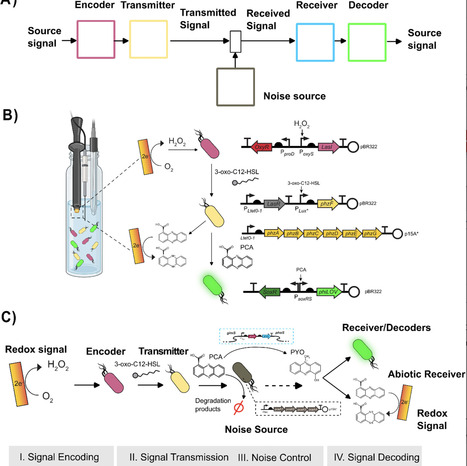
Bioelectronic systems that enable seamless communication between electronic devices and living systems represent a transformative frontier in biotechnology. Among available methodologies, redox based signaling offers unique advantages due to its ubiquity in biology and compatibility with standard electrochemical equipment, expanding on existing electrogenetic approaches while simplifying entry requirements for researchers. Here, we developed a modular phenazine-based system that enables bidirectional redox communication between electronic devices and engineered bacterial populations using commercially available electrodes. Our system integrates readily into existing synthetic biology frameworks and leverages phenazine modifications to modulate signal reception across biological and electronic domains. We structured our design around four modular components within a communication channel framework: (1) electronic signal encoding via electrochemically generated hydrogen peroxide that activates engineered cells to produce quorum sensing molecules, (2) biological signal transmission through phenazine biosynthesis controlled by a single regulatory target (PhzF), (3) dual-domain signal reception via both SoxRS-responsive biological circuits and direct electrochemical detection, and (4) controllable noise through phenazine-specific degradation enzymes. We demonstrate proportional control over phenazine production with linear relationships between electronic inputs and both biological and electrochemical outputs. This modular approach establishes phenazines as versatile bridges between electronic and biological information processing, providing accessible tools for practical bioelectronic systems with applications in environmental monitoring, adaptive biomanufacturing, and responsive biomedical devices.
|
|
Scooped by
mhryu@live.com
February 7, 3:04 PM
|
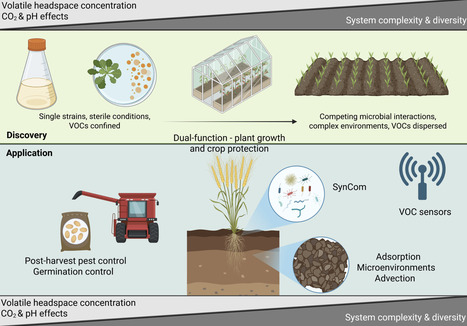
Microbial volatile organic compounds (VOCs) are integral to microbial ecological communication. Their potential as tools for sustainable crop protection is increasingly recognised, yet practical implementation remains limited. There are numerous in vitro lab-based studies focussed on screening single strains of soil or plant-associated microbes for their ability to produce VOCs and demonstrate their potential to inhibit plant pathogens or pests. Most of these, however, lack any validation in planta or in the field after petri dish experiments. This extends to a lack of understanding on whether the same VOCs are produced in vitro as in planta. How do we shift this focus and move from exciting lab-based discoveries to practical, scalable crop protection solutions for farmers? This opinion piece explores the current state of research on microbial VOCs for crop protection, translational challenges in deploying them on-farm, and highlights areas where learnings from the ecological roles of microbial VOCs can be leveraged towards field application.
|
|
Scooped by
mhryu@live.com
February 7, 2:51 PM
|
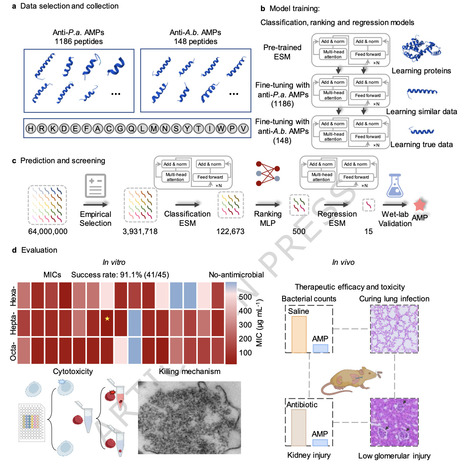
Acinetobacter baumannii, a robust Gram-negative bacterium known for causing nosocomial infections, exhibiting multidrug resistance, and lacking antimicrobial peptides that target it, remains hard to treat. Here, we report a few-shot learning pipeline integrating classification, ranking, and regression modules. Each module is trained via a few-shot learning strategy involving pre-training and multiple fine-tuning steps, incorporating similar and true data for fine-tuning, to identify potent AMP against Acinetobacter baumannii. This pipeline effectively scans complete libraries of hexapeptides, heptapeptides, and octapeptides (encompassing tens of billions of candidates) despite the extreme scarcity of training data. Results show it discovers AMPs active against Acinetobacter baumannii and Candida albicans, with low off-target toxicity and negligible drug resistance susceptibility. Additionally, EME7(7) controls Acinetobacter baumannii pneumonia in mice without kidney injury, a contrast to the observed effects of polymyxin B. This work provides a paradigm for addressing challenges of limited data availability. The authors introduce a deep learning pipeline integrating classification, ranking, and regression modules, in which each module is trained via a few-shot learning strategy involving pretraining and multiple fine-tuning steps, to identify potent AMPs against Acinetobacter baumannii.
|
|
Scooped by
mhryu@live.com
February 7, 2:42 PM
|
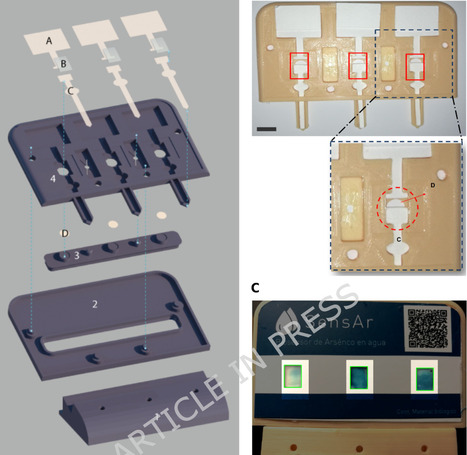
Arsenic contamination in groundwater is a critical global issue, affecting over 140 million people worldwide and posing severe public health risks, particularly in low-resource and rural communities. Argentina alone has approximately 4 million people exposed to arsenic. The measurement of arsenic in private wells is often limited by high costs, specialized personnel requirements, and geographical distances to analytical laboratories. In this paper, we describe the design and implementation of a portable, open-access arsenic biosensor that combines synthetic biology and industrial design. The biosensor employs genetically modified E. coli and a colorimetric readout to detect arsenic concentrations as low as 5 µg/L. Validation studies on 61 samples yielded a sensitivity of 98.1% and specificity of 99.0%. By using paper-based, dehydrated bacterial modules and a 3D-printed housing, this device is cost-effective, easy to use, and amenable to replication in low-resource settings. In addition, the open-access approach ensures that critical knowledge such as plasmid sequences, device schematics, and protocols can be freely shared and locally adapted. Beyond the technical advantages, this biosensor can potentially influence global policies and Argentinian programs on water quality monitoring, empowering communities to take charge of arsenic surveillance and safeguard public health.
|
|
|
Scooped by
mhryu@live.com
Today, 10:28 AM
|
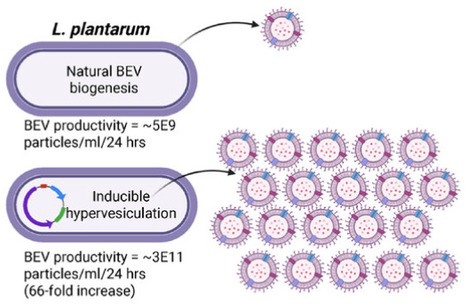
Inflammatory bowel diseases (IBD) affect over 6 million people globally and current treatments achieve only 10-20% rates of durable disease remission. Bacterial extracellular vesicles (BEVs) from probiotic lactic acid bacteria (LAB) are a promising novel therapeutic with mechanisms holding potential to drive increased rates of durable disease remission, including immunomodulation and intestinal epithelial tissue repair. However, translation of these cell-secreted nanovesicles is limited by long standing biomanufacturing hurdles, especially low production yields due to low biogenesis rates from cells. Here, Lactiplantibacillus plantarum is identified as a candidate LAB producing BEVs effective in treating acute dextran sulfate sodium (DSS)-induced murine colitis with greater efficacy than BEVs from probiotic E. coli Nissle 1917. Genetic engineering of L. plantarum to create a hypervesiculating strain via inducible expression of a peptidoglycan-modifying enzyme is shown to enable a 66-fold increase in BEV productivity. Finally, hypervesiculating L. plantarum BEVs are confirmed to be therapeutically effective in the acute DSS mouse model of colitis, with superior reduction of mucosal tissue damage compared to live L. plantarum cells. These findings demonstrate that BEVs from genetically engineered hypervesiculating strain of L. plantarum are a promising preclinical therapeutic candidate for IBD that overcomes historical biomanufacturing limitations of BEV therapeutics.
|
|
Scooped by
mhryu@live.com
Today, 10:15 AM
|
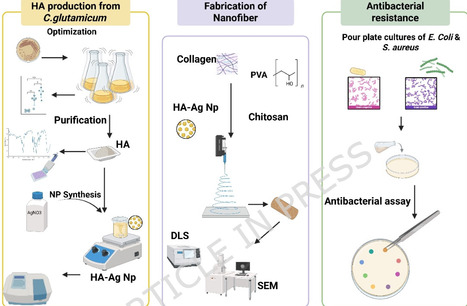
Antibiotic resistance, a growing global challenge, is predicted to cause millions of deaths in the near future. Innovative antibacterial wound dressings loaded with natural substances that have restorative effects can serve as alternatives conventional medicine. This research aims to investigate the antibacterial properties of electrospun nanofibers containing different proportions of chitosan, collagen, and hyaluronic acid-silver nanocomposite. Silver nanoparticles (Ag NPs) were synthesized via a green, solvent‑free method, with hyaluronic acid (HA) obtained from recombinant Corynebacterium glutamicum fermentation serving as the natural reducing agent. with its production optimized using a full factorial design, resulting in a 26% yield increase under conditions of 10 g/L yeast extract, 20 g/L soy protein, and 400 mg/L MgSO₄. The purity of HA obtained from microbial fermentation was measured by Fourier-Transform Infrared spectroscopy. Silver nitrate concentrations of 0.01, 0.1, and 1 M were considered to synthesize nanoparticle precursors. Spectrophotometry and Dynamic Light Scattering analyses showed that 0.1 M AgNO3 produced nanoparticles with an average size of 98.5 nm. Scanning Electron Microscopy revealed that the nanofibers had a coherent and uniform structure. Antibacterial activity was evaluated against E. coli and Staphylococcus aureus. Nanofibers with 1:1:1 and 0.5:1:1 ratios (HA-Ag: collagen: chitosan) inhibited S. aureus growth, producing inhibition zones of 1.4 cm and 1.0 cm, respectively, but showed no effect against E. coli. Cytotoxicity assessment using L929 fibroblast cells through MTT assay indicated cell viabilities of approximately 85% and 70% for the active formulations, suggesting acceptable biocompatibility. Overall, the developed nanocomposite-loaded nanofibers show potential for application against antibiotic-resistant wound infections caused by Gram-positive bacteria.
|
|
Scooped by
mhryu@live.com
February 8, 10:18 AM
|
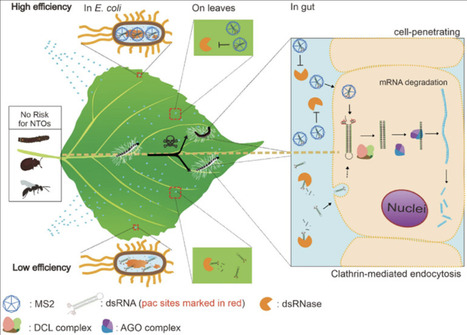
RNA interference (RNAi) represents a promising approach for insect pest management; however, its application in Lepidoptera is constrained by double-stranded RNA (dsRNA) instability, limited cellular uptake, and inefficient RNAi machinery. In this study, we developed a bacteriophage MS2 virus-like particle (VLP)-based delivery platform for hairpin RNA (hpRNA) targeting the invasive pest Hyphantria cunea. When expressed in E. coli, MS2 VLPs efficiently encapsulate hpRNA, markedly enhancing its resistance to nuclease activity and environmental degradation. In addition, surface display of the HIV trans-activator of transcription (TAT) peptide on MS2 VLPs significantly improved cellular internalization of hpRNA, resulting in robust RNAi-mediated gene silencing in H. cunea at low hpRNA doses. Importantly, no adverse effects were detected in three nontarget organisms: Clostera restitura, Plagiodera versicolora, and the parasitoid Chouioia cunea. Together, these results demonstrate that the MS2-hpRNA system represents a scalable, effective, and environmentally safe strategy for RNA-based pest control.
|
|
Scooped by
mhryu@live.com
February 8, 10:07 AM
|
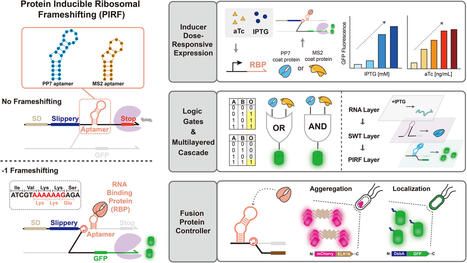
Programmed ribosomal frameshifting (PRF) is a translational mechanism that enables the ribosome to shift reading frames and access alternative coding sequences. PRF occurs naturally in a wide range of organisms, including viruses, bacteria, and eukaryotes, where it supports compact encoding and stoichiometric control of protein expression. Despite the great potential of PRF in synthetic circuit designs, a broader adoption of PRF in circuit designs has been hampered by rather strict sequence constraints and structural requirements. This work introduces a synthetic translational regulatory platform, protein-inducible ribosomal frameshifting (PIRF), by integrating aptamer–protein interactions with a − 1 PRF motif to enable regulated translation in E. coli. PIRF modules respond to intracellular RNA-binding proteins such as PP7 and MS2, triggering frameshifting in a condition-dependent manner. PIRF could be used to program logic gate operations through frame-dependent translation and enable multilayered regulation in synthetic circuits. Further, the flexible PIRF designs enable reading frame-dependent control of fusion protein expression, protein aggregation, and periplasmic localization via strategic positioning of peptide tags and protein coding sequences. While PIRF enabled regulated frameshifting and could be flexibly reconfigured for a variety of circuits and applications, a measurable level of basal frameshifting was often observed, which may require additional strategies for further optimization in the future. Together, PIRF supports applications in programmable and logical control of downstream protein expression, including condition-dependent aggregation and regulated subcellular localization. PIRF provides a compact and genetically encoded strategy for programmable protein-level regulation, expanding the synthetic biology toolkit for translational control, biosensing and biotherapeutics.
|
|
Scooped by
mhryu@live.com
February 8, 1:01 AM
|
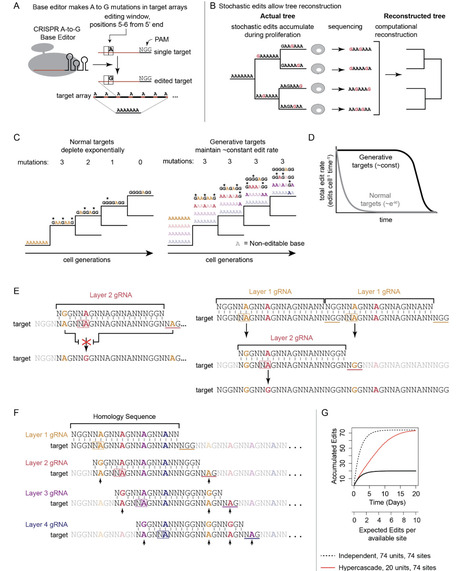
Reconstructing the lineage histories of individual cells can reveal the dynamics of developmental and disease processes. In engineered recording systems, cells stochastically edit synthetic barcode sequences as they proliferate, creating distinct, heritable edit patterns that can be used to reconstruct the lineage trees relating individual cells in a manner analogous to phylogenetic reconstruction. However, recording depth is often limited by the kinetics of the editing process: the rate of editing declines exponentially over time for an array of independently editable targets, leading to most edits occurring in early generations. Here, we introduce the hypercascade, a regenerative molecular recording system that takes advantage of the predictability of A-to-G base editing to progressively create new target sites over time. The hypercascade packs 4 editable target sites in every 20 bp of sequence, enabling high density information storage. More importantly, the hypercascade's regenerative logic leads to an approximately constant rate of mutation accumulation over time. This in turn facilitates reconstruction of deep lineage relationships. We demonstrate this by reconstructing trees spanning 23 days of editing and approximately 17 generations after a single polyclonal engineering step. Finally, simulations show that the hypercascade has the potential to record chromatin state transition dynamics across multiple genomic loci in parallel. The hypercascade thus provides a flexible and broadly useful tool for molecular recording.
|
|
Scooped by
mhryu@live.com
February 8, 12:37 AM
|
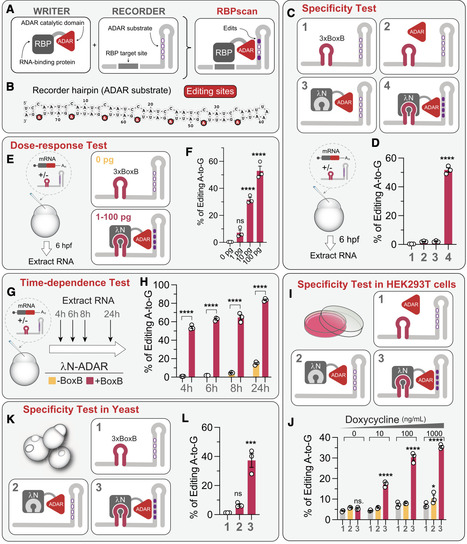
RNA-binding proteins (RBPs) are essential regulators of gene expression at the post-transcriptional level, yet obtaining quantitative insights into RBP-RNA interactions in vivo remains challenging. Here, we developed RBP specificity and contextual analysis via nucleotide editing (RBPscan), which integrates RNA editing with massively parallel reporter assays to profile RBP binding in vivo. In RBPscan, fusion of an RBP to the adenosine deaminase acting on RNA (ADAR) catalytic domain induces RNA editing of a recorder mRNA carrying the tested RBP-binding site, serving as a readout of the RBP-RNA interaction. We demonstrate the utility of RBPscan in zebrafish embryos, human cells, and yeast, showing that it quantifies binding strength, resolves dissociation constants, identifies binding motifs for various RBPs, and links binding affinities to their impact on mRNA stability. RBPscan also provides positional mapping of Pumilio-binding sites in the long non-coding RNA NORAD. With its simplicity, scalability, and cross-system compatibility, RBPscan is a versatile tool for investigating protein-RNA interactions and complements established methods for studying post-transcriptional regulatory networks.
|
|
Scooped by
mhryu@live.com
February 8, 12:29 AM
|
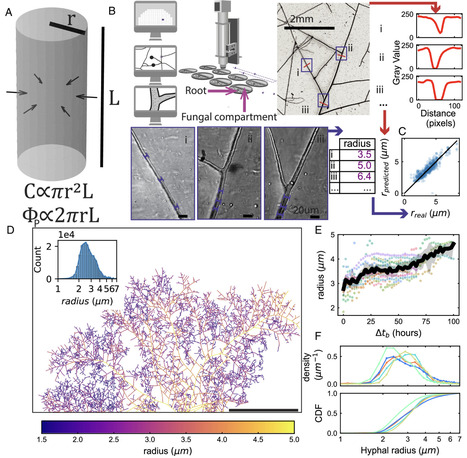
Symbiotic nutrient exchange between arbuscular mycorrhizal (AM) fungi and their host plants varies widely depending on their physical, chemical, and biological environment. Yet dissecting this context dependency remains challenging because we lack methods for tracking nutrients such as carbon (C) and phosphorus (P). Here, we developed an approach to quantitatively estimate C and P fluxes in the AMF symbiosis from comprehensive network morphology quantification, achieved by robotic imaging and machine learning based on roughly 100 million hyphal shape measurements. We found that rates of C transfer from the plant and P transfer from the fungus were, on average, related proportionally to one another. This ratio was nearly invariant across AMF strains despite contrasting growth phenotypes but was strongly affected by plant host genotype. Fungal phenotype distributions were bounded by a Pareto front with a shape favoring specialization in an exploration–exploitation trade-off. This means AMF can be fast range expanders or fast resource extractors, but not both. Manipulating the C/P exchange rate by swapping the plant host genotype shifted this Pareto front, indicating that the exchange rate constrains possible AMF growth strategies. We show by mathematical modeling how AMF growth at fixed exchange rate leads to qualitatively different symbiotic outcomes depending on fungal traits and nutrient availability.
|
|
Scooped by
mhryu@live.com
February 8, 12:16 AM
|
Antisense oligonucleotides (ASOs) are short, synthetic nucleic acids that bind to complementary RNA sequences and alter gene expression, making them versatile therapeutic agents. Here, we identify transcription termination windows of protein-coding genes as previously unrecognized targets for ASOs. We show that ASOs can act on nascent RNA synthesised downstream of the annotated genes, leading to a pronounced decrease in the corresponding mRNA levels. These downstream-of-gene ASOs (DG-ASOs) induce RNase H1-dependent cleavage, impairing mRNA 3′ end processing, directing the unprocessed mRNAs for exosome-dependent degradation and creating early entry points in the nascent RNA for the XRN2 exonuclease termination factor. Altogether, we show that termination windows are genuine ASO targets which can be exploited to suppress gene expression. Importantly, we also reveal an underappreciated source of off-target effects which may arise from ASOs binding downstream of genes. Our findings indicate the necessity to expand ASO design and off-target assessment guidelines to include termination window sequences, thereby improving therapeutic efficacy and safety.
|
|
Scooped by
mhryu@live.com
February 8, 12:00 AM
|
Bacillus subtilis is widely employed for lignocellulose degradation. However, wild-type strains typically exhibit low and incomplete cellulase activities. This study aimed to engineer recombinant B. subtilis strains harboring complete, high-efficiency cellulase systems using CRISPR/Cas9-mediated genome editing. After optimization of signal peptides, transcriptional terminators, and chromosomal integration sites, two endoglucanase expression cassettes were integrated into B. subtilis 168, yielding a strain with a 16.29-fold increase in endoglucanase activity relative to the parental strain. In parallel, a bifunctional cellulase was successfully expressed and optimized, achieving exoglucanase (549.77 U/mL) and β-glucosidase (349.26 U/mL) activities. Moreover, the strain BSKI3Cel, containing three optimized expression cassettes in its genome, exhibited high endoglucanase (129.59 U/mL), exoglucanase (596.75 U/mL), and β-glucosidase (447.42 U/mL) activities. Subsequent transformation of BSKI3Cel with plasmid pJOE2006Bf, carrying genes encoding a composite cellulase system, yielded BSK3P2C, which achieved peak cellulase activities of 533.16, 2959.83, and 2829.61 U/mL on day 8. As a proof of concept, fermentation of wheat straw using BSK3P2C was found to significantly reduce hemicellulose (16.70 %), neutral detergent fiber (7.46 %) and acid detergent fiber (9.93 %) contents. Microscopic analyses confirmed extensive lignocellulose degradation. Overall, this study establishes a high-performance B. subtilis platform with complete cellulase systems for efficient cellulosic biomass conversion.
|
|
Scooped by
mhryu@live.com
February 7, 3:10 PM
|
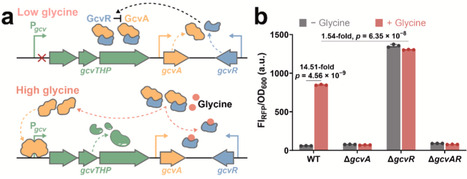
Glycine is an essential metabolite and a valuable precursor for various industrially relevant compounds. However, the lack of efficient high-throughput detection methods has limited progress in metabolic engineering for glycine bioproduction. In this study, we developed a transcription factor-based glycine biosensor in E. coli by reprogramming the native GcvA/GcvR regulatory complex. By fine-tuning the expression balance between GcvA and GcvR, the biosensor exhibited significantly reduced basal fluorescence while achieving strong glycine-dependent induction of up to 16.45-fold. The optimized system demonstrated rapid response kinetics, high sensitivity, and excellent specificity toward exogenously supplied glycine. The sensor accurately reflected intracellular glycine concentrations in engineered glycine-producing strains, with fluorescence output correlating with production levels. Furthermore, its application enabled high-throughput screening of SerC (phosphoserine aminotransferase) mutants with enhanced glycine production. The best-performing SerC variant resulted in a 48.54-fold increase in fluorescence intensity and a 2.25-fold improvement in glycine accumulation, validating the biosensor as an effective tool for directed enzyme evolution. This work established a robust and tunable platform for glycine biosensing, offering broad potential for metabolic pathway optimization and enzyme engineering related to glycine metabolism.
|
|
Scooped by
mhryu@live.com
February 7, 2:56 PM
|
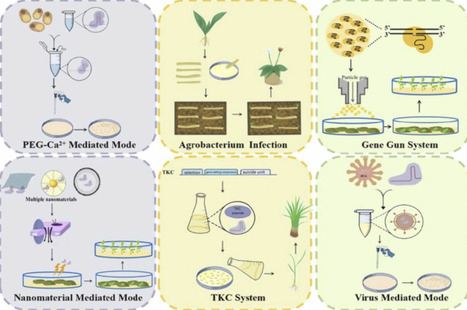
The rapid advancement of CRISPR/Cas-mediated genome editing has revolutionized plant biotechnology, yet the integration of exogenous DNA into plant genomes raises biosafety concerns and regulatory hurdles. Transgene-free genome editing technologies, which eliminate foreign gene remnants while enabling precise modifications, are critical for the commercialization and ecological sustainability of edited plants. This review provides a comprehensive and integrated analysis of transgene-free editing strategies, focusing on the latest advances in three core innovations: (1) Optimization of ribonucleoprotein (RNP) components: A systematic side-by-side comparison of editing efficiencies between Cas variants and gRNA variants—enhancing editing specificity, reducing off-target effects, and eliminating transgene integration; (2) Delivery systems, including PEG-Ca²⁺ mediated, particle bombardment delivery and nanomaterial-based platforms, which enable transgene-free of CRISPR components while bypassing tissue culture; (3) gene module molecular toolkits, including high-frequency regeneration modules, negative selection module and visualization module, which represent an underexplored frontier in previous reviews. By integrating these innovations, transgene-free editing technologies hold immense potential for perennial plants, enabling trait improvements in yield, stress tolerance, and disease resistance without compromising genetic integrity. This review highlights remaining challenges, including delivery efficiency in recalcitrant species and scalability for high-throughput applications, while underscoring the role of artificial intelligence and machine learning in advancing next-generation editing tools. This work not only synthesizes key technological advances but also provides a clear roadmap for addressing challenges related to delivery efficiency, regulatory hurdles, and public acceptance, thereby paving the way for sustainable agriculture and the global adoption of CRISPR-edited plants.
|
|
Scooped by
mhryu@live.com
February 7, 2:48 PM
|
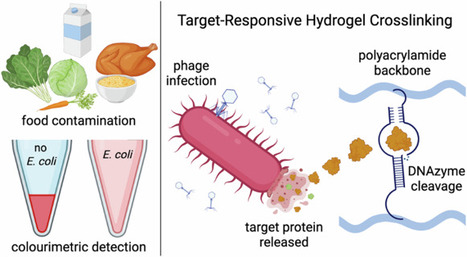
Due to the significant healthcare burden associated with foodborne illness, developing platforms suitable for the on-site detection of food pathogens is of critical importance to public health. Low-cost, equipment-free approaches are desired to allow for point-of-use contamination monitoring along the food supply chain. Here, we demonstrate the compatibility of an E. coli responsive colorimetric DNAzyme-crosslinked hydrogel sensor with a wide range of food products. Sensor functionality involves an E. coli detecting DNAzyme-substrate complex that cleaves the hydrogel crosslinking in the presence of the target bacteria, resulting in a release of gold nanoparticles that is visible to the naked eye. Naked-eye detection of E. coli at concentrations of 105 CFU mL-1 has been shown in milk as well as samples extracted from produce, leafy greens, and ready-to-eat foods such as rotisserie chickens. The functionality, simplicity, and versatility of this sensing platform may improve the feasibility of frequent pathogen monitoring in the food production pipeline, with the potential to mitigate future outbreaks of foodborne illness.
|

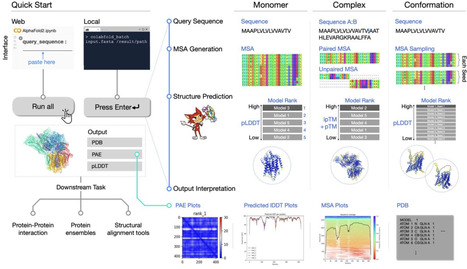



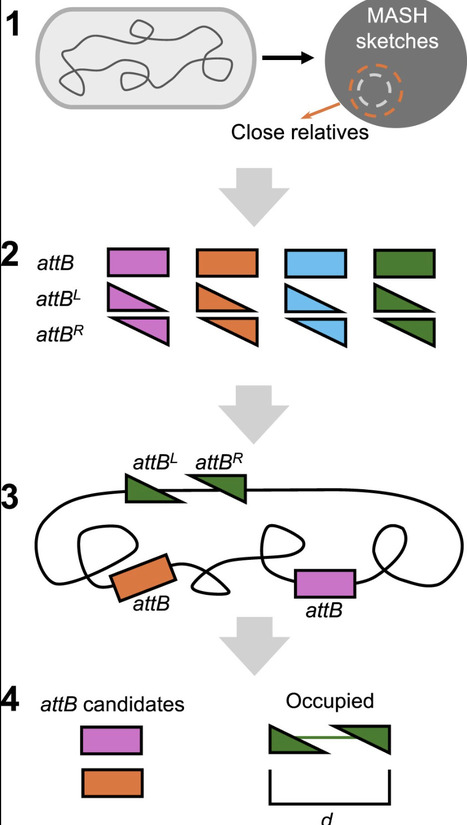
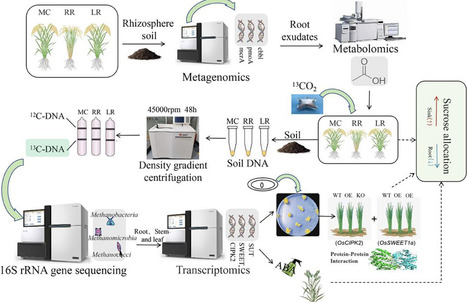








af2 protocol, steinegger, 3st, now npc https://www.nature.com/articles/s41596-024-01060-5