 Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 11:05 AM
|
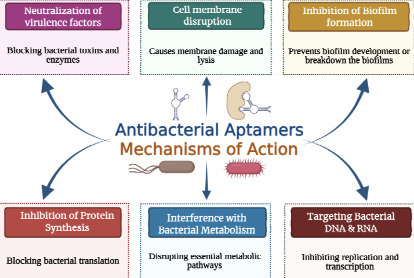
In recent years, aptamers have transitioned from mere laboratory tools to highly potent molecular recognition agents capable of overcoming the strict limitations of conventional antibiotic therapies. We have developed AptBacterialDB, a manually curated, large, comprehensive database of experimentally validated antibacterial aptamers spanning 1996 to 2026. The database contains a total of 2131 aptamers targeting approx 75 different bacterial classes, and 124 aptamer targets with 95 entries found in UTexas databases, 97 in AptaDB, and 28 in Aptabase. It contains 1555 unique aptamer sequences, 189 unique modifications, 40 different selection approaches, and 44 different affinity methods. It integrates detailed annotations of about 20 fields, including sequence information, nucleic acid type, binding affinity, modifications, experimental and functional details. The secondary structure of the aptamers was predicted using ViennaRNA Package 2.0, demonstrating that they adopt mostly stable conformations, with a structured stem region. MySQL was implemented for database development, and a knowledge graph was integrated using ArcadeDB/openCypher for graphical visualization of aptamer-target-organisation relationships. Facilities such as different search modes, browsing, similarity search, REST API access, and entries linked to the existing database for a broader view of the aptamers have been provided. AptBacterialDB (https://webs.iiitd.edu.in/raghava/aptbacterialdb/) provides a user-friendly centralized platform to accelerate antibacterial aptamer research, therapeutic development, biosensor design, and computational modelling efforts.
|
|
Scooped by
mhryu@live.com
Today, 10:55 AM
|
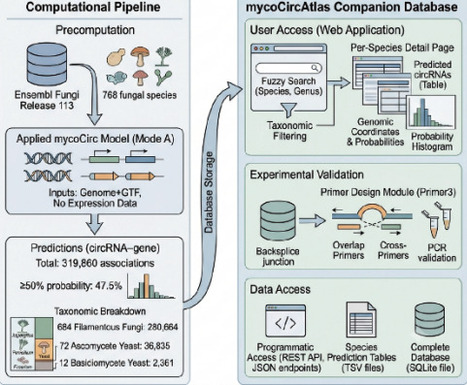
Exploring the fungal circular RNA (circRNA) landscape is bottlenecked by both experimental and computational limits. While standard mRNA-seq systematically discards circRNAs due to their lack of poly(A) tails, high-cost total RNA-seq remains prohibitive for large-scale screening. Consequently, discovery relies heavily on computational prediction. However, existing models trained exclusively on human or plant sequences fail in fungi because of the extreme genomic divergence across fungal lineages, which span from intron-poor Candida to intron-rich filamentous fungi. As a result, no computational framework currently exists for de novo fungal circRNA prediction, leaving the vast majority of non-model fungi entirely inaccessible. Results: We present mycoCirc, the first end-to-end pan-fungal multi-modal pretrained model for fungal circRNA prediction, integrating three mandatory modalities with bidirectional cross-attention for donor-acceptor site interaction. Pre-trained on 22 strains with 16,483 positive gene-circRNA associations spanning Ascomycete yeast, Basidiomycete yeast, and Filamentous fungi groups and fine-tuned per group using 5-fold cross-validation, mycoCirc achieved AUROC 0.69-0.70 on held-out test species under Mode A (Genome+GTF, no RNA-seq), substantially outperforming JEDI (0.51-0.57) and CircPCBL (0.49-0.53). Cross-species evaluation on four independent fungi datasets demonstrated robust generalization across all fine-tuned variants (AUROC 0.63-0.72). Beyond gene-level classification, the JunctionEncoder module enabled backsplicing junction identification for detailed circRNA validation. We further build mycoCircAtlas, a companion database providing 319,860 high-confidence gene-circRNA predictions across 768 fungal species from Ensembl Fungi Release 113, enabling researchers to query precomputed predictions and design validation primers without local model deployment.
|
|
Scooped by
mhryu@live.com
July 3, 4:36 PM
|
Microbial communities carry out important ecological functions. Their activities emerge from interactions between species, often potentiated by metabolic traits. We lack a quantitative understanding of how these traits shape community properties. Here, we present theory for microbial communities, leveraging concepts from quantitative microbial physiology. We focus on how steady-state metabolic exchanges between species determine their fractional abundances, given their biomass and byproduct yields on nutrients. We start by deriving formal conditions for the steady states of communities of microbes that grow, die and cross-feed metabolites. We describe the metabolic stoichiometry of nutrient uptake and the formation of biomass and byproducts for each species in terms of charge- and chemical-element balanced reactions (macrochemical reactions). Byproducts function as nutrients for other species. Next, we express the relative abundances of species (living and dead), the net metabolic conversion of a community, and the biomass carrying capacity in terms of the metabolic stoichiometry, growth rates and death rates of the species. We show how niche creation can emerge from stoichiometric imbalances in cross-feeding communities. Finally, we discuss how relative species abundances depend on the ATP stoichiometries of intracellular metabolism.
|
|
Scooped by
mhryu@live.com
July 3, 4:22 PM
|
Bacterial pathogens rely on the constant availability of purine and pyrimidine nucleotides to facilitate replication, growth, and virulence and to sustain energy metabolism and nucleotide-based signaling. The capacity to switch between de novo synthesis and salvage pathways underpins much of their metabolic flexibility and also regulates access to different human body niches, where nucleobase availability varies significantly between extracellular fluids, mucosal surfaces, inflamed tissues, and intracellular compartments. However, adaptation to specific host niches can result in the loss of de novo nucleotide biosynthesis pathways, increasing bacterial dependence on nucleobase/nucleoside salvage. Many intracellular pathogens lack de novo synthesis pathways, making purine or pyrimidine salvage not an optional, but an essential process where host nucleotide reserves are critical to bacterial survival. Because of their central role in bacterial metabolism, enzymes, transporters, and regulatory networks involved in purine and pyrimidine metabolism represent potential targets for therapeutic interventions. This review summarizes the current knowledge of purine and pyrimidine metabolism in bacterial pathogens, including the abundance of these compounds in different host niches, tissue-specific fitness strategies, and bacterial targets for further development of innovative antibacterials.
|
|
Scooped by
mhryu@live.com
July 3, 4:08 PM
|
Microbial communities in natural environments are consistently challenged by toxic compounds, such as polycyclic aromatic hydrocarbons (PAHs). The capacity for rapid phenotypic diversification is key to survival in these dynamic and often hostile niches. Here, we report a mechanism by which E. coli generates immediate phenotypic heterogeneity in response to a model PAH, perylene. Utilizing single-cell techniques, we revealed unexpected efflux phenotypic switches occurring in a single division event by examining the asymmetric distribution of perylene, a TolC-related efflux system substrate. Contrary to gradual changes in many generations, the phenotypic switch we observed is abrupt, resulting in daughter cells with distinct efflux phenotypes and asymmetric viability. We ruled out uneven efflux pump distribution and suggested the switch is linked to asymmetric proton motive force. Furthermore, we found a correlation between the age of the inherited pole during division and the propensity for phenotypic switching. New pole cells are identified as more prone to this switch. Our findings reveal a rapid division-based strategy for generating phenotypic diversity that could enhance the adaptive potential and resilience of bacterial populations facing fluctuating environmental toxins.
|
|
Scooped by
mhryu@live.com
July 3, 3:46 PM
|
Phaeobacter inhibens T5T harbors three LuxI/LuxR quorum-sensing systems: one encoded by the host and one in each of two prophages. Each prophage quorum-sensing autoinducer activates its own lysogeny program and that of the co-resident prophage. By driving competitors toward lysogeny, prophages deny their rivals access to susceptible hosts. One prophage engages in a bidirectional interaction with the host: the prophage activates host quorum sensing, while the host represses prophage quorum sensing. Because the host and prophage autoinducers exert opposing effects on the same prophage promoter, the prophage's lysis-lysogeny decision depends on the ratio of the two autoinducers rather than their absolute concentrations. Ratiometric sensing allows the prophage to infer the relative abundances of infected and uninfected hosts and to commit to lysis only when uninfected hosts predominate. Phage quorum-sensing modules are widespread and undergo diversification and exchange. These findings reveal how prophages surveil and manipulate competitors that share chemical environments.
|
|
Scooped by
mhryu@live.com
July 3, 3:25 PM
|
Artificial metalloenzymes (ArMs) are proteins engineered to contain metal cofactors that often catalyze chemical reactions rarely or never observed in natural biological processes. They hold promise for applications including fine-chemical production, control of cellular function and therapeutics. Many of these applications are difficult to achieve due to cofactor inactivation in complex biological environments and cofactor-induced cellular stress. In this study, I explore the most recent strategies for developing robust, biocompatible ArMs that function in cell lysates, on cell surfaces or intracellularly. The pros and cons of developing and using ArMs in these three environments are described. I also examine how active ArMs might tolerate their environment, and the outstanding challenges and opportunities, including the need for simple methods of construction, improved catalytic performance and exploration of other reactions and microorganisms. Bringing transition-metal catalysis into biological systems remains challenging. Here the author examines how artificial metalloenzymes can be made to function across biological settings, from cell extracts to cell surfaces and intracellular environments.
|
|
Scooped by
mhryu@live.com
July 3, 2:57 PM
|
To address emerging climate and resource threats to global agriculture, we require advanced, plant-to-farm monitoring interventions. This perspective proposes Flora-Fi, an expanded Internet of plants framework leveraging innate intra- and interplant communication (IPC) pathways. By integrating biological signals with digital networks, Flora-Fi enables energy-efficient, early stress detection. We outline a blueprint detailing the next-generation sensing, communication, and data processing infrastructures necessary to realize this holistic crop management paradigm.
|
|
Scooped by
mhryu@live.com
July 3, 12:47 AM
|
Plants have long served as natural indicators of environmental conditions, and recent advances in synthetic biology are enabling the design of engineered sentinels — living sensors that can report on abiotic and biotic stressors. This review summarizes recent advances in designing sensor plants, also called phytosensors or sentinel plants, highlighting three major strategies: (1) exploiting native promoter systems responsive to environmental cues, (2) engineering protein-based genetically encoded biosensors that detect specific molecules of interest, and (3) constructing interkingdom signaling networks between plants and microbes to extend sensing capabilities to the rhizosphere. These sense-response modules can be coupled to optical reporters (e.g., fluorescence, bioluminescence, and pigment-based) that enable remote detection via drones and satellite imaging. Continued improvements in promoter design, receptor modularity, and signal visualization technologies are driving the development of robust, field-deployable plant biosensors. Together, these innovations position engineered sensor plants as scalable, self-sustaining sentinels for real-time environmental monitoring and land management.
|
|
Scooped by
mhryu@live.com
July 2, 11:52 PM
|
Nanopore direct RNA sequencing (DRS) has transformed transcriptomics by enabling single-molecule, long-read sequencing of native RNA without the need for reverse transcription or amplification. In contrast to short-read RNA-seq and cDNA-based long-read approaches, DRS can simultaneously capture multiple RNA modifications, full-length transcript architecture, alternative splicing patterns, and poly(A) tail features within individual molecules, thereby providing an integrated view of transcriptomic and epitranscriptomic regulation. In this comprehensive review, we outline the biophysical principles underlying nanopore DRS and trace its technological evolution. We compare its performance with short-read RNA sequencing, long-read cDNA sequencing, and conventional RNA-modification mapping strategies, highlighting its advantages in isoform-resolved quantification and multilayer RNA feature integration, while also clarifying contexts in which alternative or combined approaches may be more appropriate for robust biological interpretation. We further summarize optimized experimental workflows, including library construction strategies tailored to diverse RNA biotypes (mRNA, rRNA, tRNA, circRNA, miRNA, and nonpoly(A) transcripts), as well as recommended quality-control procedures and sequencing optimization practices. Emphasizing recent computational advances and translational applications of DRS, we cover state-of-the-art algorithms for RNA modification detection, transcript reconstruction, and isoform quantification. We also propose analytical pipelines for poly(A) tail length inference and integrative frameworks that jointly analyze these regulatory layers. We distinguish direct nanopore signals from computational inferences to define confidence levels and emphasize benchmarking and orthogonal validation of readouts. Practical implementation examples are included to facilitate reproducible analysis. Finally, we highlight emerging applications of integrated DRS, including the resolution of complex transcriptomes, the characterization of coordinated epitranscriptomic regulation, and the identification of disease-associated RNA signatures. We also discuss current technical challenges and future perspectives, particularly in relation to multi-omics integration and the broader deployment of DRS in precision medicine as well as in plant and animal research.
|
|
Scooped by
mhryu@live.com
July 2, 11:34 PM
|
Artificial intelligence (AI) strategies are revolutionizing genomics by extracting complex patterns that traditional statistical pipelines are likely to miss. This mini-review aims to provide a concise overview of how AI is transforming major genomic technologies including variant calling, gene expression analysis, single-cell transcriptomics, CRISPR-Cas9 optimization, and multi-omics integration. In genome sequencing, machine learning variant callers greatly improve the accuracy and the rate at which single nucleotide and structural variants are called. In bulk RNA-Seq, AI augmented quantification, denoising, and differential expression modules complement the highly established STAR-featureCounts-DESeq2 pipeline, revealing subtle signals in big data sets. In single cell transcriptomics, deep learning approaches enhance batch correction, automate cell type annotation, and track developmental trajectories, hence clarifying cellular heterogeneity. AI-assisted guide RNA design, outcome prediction, and nuclease engineering enable more efficient CRISPR-Cas9 editing, reducing experimental cycles, and off-target effects. Finally, integrated platforms that combine genomic, transcriptomic, epigenomic, proteomic, and metabolomic layers provide an integrative view of cellular regulation and disease mechanisms. The review also covers current limitations, sparsity of data, model bias, privacy, and the need for standardized benchmarks and offers future directions in the form of interpretable models, collaborative learning, and open science practices. Together, these developments render AI an indispensable partner to unravel genomic complexity and accelerate precision medicine applications.
|
|
Scooped by
mhryu@live.com
July 2, 11:28 PM
|
CRISPR-Cas9 genome-editing efficiency is strongly influenced by the sequence composition and positional context of single-guide RNAs (sgRNAs). Although numerous deep learning–based models have been developed to predict Cas9 efficiency from sgRNA sequences, most operate as black boxes, offering limited insight into the sequence determinants underlying Cas9 activity. In addition, previous studies often overlook how the positional context of sequence motifs within sgRNAs influences their effects on Cas9 binding or cleavage. We introduce DeepCC9, an interpretable machine learning framework that combines explicit sequence feature extraction with a residual block–based deep architecture to improve interpretability and identify composition- and position-based motifs governing Cas9 genome-editing efficiency. We applied this method to multiple Cas9 variant datasets, achieving superior predictive performance compared with existing methods while enabling direct interpretation of sequence motifs and their positional effects. Our analysis uncovered 74 sequence motifs enriched or depleted at specific positions within sgRNAs and strongly associated with Cas9 efficiency, providing mechanistic insight into sequence features that influence guide performance. Together, these results establish DeepCC9 as a generalizable and interpretable framework for modeling sequence–function relationships and advancing the understanding of the sequence determinants underlying CRISPR-Cas9 genome editing.
|
|
Scooped by
mhryu@live.com
July 2, 10:49 PM
|
Lichens are symbiotic associations between a fungal mycobiont and a photosynthetic photobiont. They thrive in nutrient-poor environments; yet the mechanisms underlying their adaptation to iron limitation remained largely unknown. Here, we characterize the iron acquisition system of Xanthoria parietina, a globally distributed lichen-forming fungus associated with the microalgal photobiont Trebouxia decolorans. We demonstrate that the mycobiont produces the siderophore ferrichrome and possesses the full genetic repertoire not only for siderophore biosynthesis, but also reductive iron assimilation, iron detoxification, and regulation. The ferrichrome-synthesizing non-ribosomal peptides synthetase exhibits a lichen-specific compact architecture but retains functionality when heterologously expressed in a non-lichenized ascomycete. Transcriptomic analysis and ferrichrome quantification reveal substrate-dependent regulation of the siderophore system. Importantly, ferrichrome promotes photobiont growth independent of extracellular iron reduction, indicating direct utilization. These findings provide the functional evidence of siderophore-mediated iron acquisition in a lichen symbiosis and highlight ferrichrome as a key mediator of mutualistic nutrient exchange. The mechanisms underlying the adaptation of lichens to nutrient-poor environments are poorly understood. Here, Happacher et al. show that a globally distributed lichen fungus produces an iron-scavenging molecule that promotes growth of its algal partner.
|
|
|
Scooped by
mhryu@live.com
Today, 11:03 AM
|
DNA foundation models are trained to predict the likelihood of natural sequences, but the calibration between such likelihood scores and laboratory fitness or directly measured molecular phenotypes depends strongly on gene context, sequence divergence from wild-type, and selection regime. We apply zero-shot variant scoring with Evo2 7B (dLLR, the change in pseudo-log-likelihood between mutant and reference windows) to five E. coli datasets and quantify this context-dependent calibration map. Calibration is strong in two settings. In the Firnberg 2014 deep mutational scan of TEM-1 beta-lactamase (13,027 nucleotide-level variants; plasmid-borne enzyme under band-pass ampicillin selection), Evo2 dLLR tracks measured fitness at Spearman rho = 0.545 (95% CI 0.532-0.557; SNV rho = 0.606, indel rho = 0.521). In the Tenaillon 2012 thermal-evolution dataset, type-stratified, window-tuned scoring reaches Insertion AUROC 0.882 (W = 2,048 bp) and Deletion AUROC 0.846 (W = 4,096 bp). Calibration is decisively absent in the same organism: the Ireland 2020 RegSeq promoter MPRA gives rho = 0.011 (95% CI 0.003-0.019; n = 64,665), flat even after -10/-35 mechanism stratification, and the Dewachter 2023 chromosomal-essentials scan (fabZ/lpxC/murA) gives rho = 0.041 (95% CI 0.025-0.058). The Papkou 2023 folA combinatorial landscape sits between, at rho = 0.237, with a sweep that falls monotonically from rho = 0.575 at two mutations from wild-type to rho = 0.065 at nine. Pooling per-gene and per-divergence correlations, we fit calibration as an explicit function rho = f(sequence divergence from WT, variant context): weighted regression gives a negative divergence coefficient and a negative regulatory-context coefficient (both in the predicted direction; R2 = 0.49), an explicit, if illustrative, fit rather than a metaphor. We further test, and find unsupported, the intuitive explanation for the residual TEM-1 vs. essentials gap: across five genes the chromosomal essentials are more represented than TEM-1 by raw public-database deposition couno simple training over-representation does not exiant diversity is a candidate but remains untested. We therefore reframe Evo2 not a a likelihood predictor whosecalibration with fitness is conble is not a DMS pre-screen toolbut a quantitative lookup table likelihood-fitness gap closes(training-rich plasmid CDS undeens (chromosomal essentials,native promoter regulatory variorganism, plasmid vs. chromosomal context and strong vs. weak selifferent calibration regimes,the central finding.
|
|
Scooped by
mhryu@live.com
Today, 10:52 AM
|
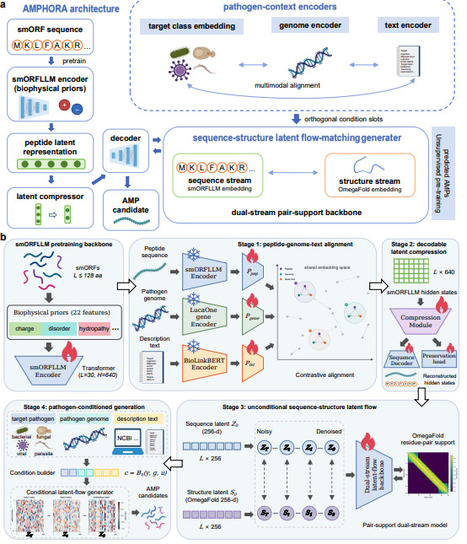
Antimicrobial peptide discovery is constrained less by the number of molecules that can be generated than by the choice of which few should be tested against a defined pathogen. Most peptide generators produce broadly antimicrobial-like sequences and leave target specificity to downstream filters. Here we show that pathogen context can be introduced during generation. AMPHORA conditions a peptide-native generator on target class, pathogen genome features and strain-description text. Matched, ablated and shuffled controls showed that aligned pathogen inputs redirected generated libraries beyond coarse activity labels, whereas global shuffling weakened this effect. Same-noise counterfactuals showed that strain descriptions drove larger sequence changes, whereas genome features more strongly affected predicted structural properties. Species-level analyses revealed target-dependent enrichment. Matched bacterial inputs also shifted APEX-predicted activity rankings relative to class-only generation. The resulting libraries remained diverse, largely non-memorizing and compatible with predicted peptide-like structural features. Together, these results establish pathogen-context conditioning as a new paradigm for computational library reshaping in antimicrobial peptide generation.
|
|
Scooped by
mhryu@live.com
July 3, 4:30 PM
|
Insertion sequences (IS) are widely involved in bacterial genomic plasticity by disrupting, adding, moving genomic sequences, or by activating or extinguishing gene expression. A specific family of IS, ISCR (for insertion sequence of Common Region), is thought to be involved in the dissemination of antibiotic resistance genes (ARGs). While some ISCR members are commonly found in bacteria isolated in clinical settings and can contribute to downstream ARG expression, the mechanisms regulating the ISCR-encoded transposase expression have remained uncharacterized. Here, we investigated the expression of the transposase genes of ISCR1, ISCR2, and ISCR8 and its regulation in E. coli. Using in silico analyses and in vitro experiments, we showed that the expression levels were extremely low, as observed for most IS transposases. We further demonstrated the direct role of DNA damages and the key SOS response repressor, LexA, in controlling the activity of the transposase promoter. These results provide evidence that the mobility of at least some ISCR elements may be promoted upon bacterial exposure to antibiotics inducing the SOS response.
|
|
Scooped by
mhryu@live.com
July 3, 4:18 PM
|
The high cost of cultivation media remains one of the barriers to the commercialization of cultivated meat. Current serum-free media reduce reliance on fetal bovine serum but only achieve modest cost savings. Especially at larger scales (> 20 m3), pharmaceutical-grade amino acids and recombinant proteins remain major cost drivers. Techno-economic analyses suggest that replacing purified amino acids is essential to achieve price parity with conventional meat. Microbial hydrolysates, also referred to as extracts, offer a promising alternative, as these can supply complex mixtures of amino acids and peptides at lower cost and with greater scalability than chemically defined formulations. In particular, yeast and bacterial extracts combine high protein (and therefore high amino acid) content, rapid growth, and established industrial-scale production. Comparative analyses indicate that microbial amino acid profiles broadly overlap with those of animal cells and traditional meat, but this similarity does not translate into meeting cellular amino acid demand. Consumption data indicate that key amino acids, such as glutamine, cysteine, serine and arginine, would be supplied insufficiently by microbial extracts. This mismatch highlights the need for engineering of microbial biomass and optimization of extraction methods. Extraction methods such as autolysis, enzymatic lysis, or physical disruption strongly influence nutrient release and composition, underscoring the need for standardizing microbial extract preparation. In addition, challenges remain in ensuring consistency, safety, and bioavailability of nutrients, as microbial extracts also contain nucleic acids and potential toxins. This review summarizes current knowledge on microbial hydrolysates for cultivated meat media, including biomass composition, extraction methods, and analytical tools to assess quality and performance. We identify key knowledge gaps, particularly in quantitative amino acid consumption data for relevant cell lines and performance testing in scalable cultures. Addressing these gaps could enable cost-effective media development and accelerate research toward sustainable, commercially viable cultivated meat.
|
|
Scooped by
mhryu@live.com
July 3, 3:51 PM
|
Phage therapy offers a promising solution to the antimicrobial resistance crisis. However, a major concern preventing the adoption of phage therapy is the potential for unintended consequences of phage release; both in regard to preventing the spread of phage resistance, and the proliferation of a non-endemic virus into the microbial ecosystem. Conditional replication (biocontainment) of phages through bioengineering may address these concerns, but the impact on bactericidal efficacy is unknown. Here, we created a biocontained T7 phage (T7Δcapsid) lacking the major structural capsid gene, gp10AB, that can only replicate on Escherichia coli strains expressing gp10AB in trans, and assessed its bactericidal efficacy compared with wild-type T7. Congruent with model predictions, T7Δcapsid was only able to clear a well-mixed culture of E. coli at a multiplicity of infection (MOI) of 10 or higher, whereas wild-type T7 prohibited growth at an MOI of 0.1. The reduction in efficacy was more evident in a complex structured environment within a microfluidic device, where phage success depends on its ability to penetrate a microbial niche via propagation. In this environment, T7Δcapsid was unable to propagate into the bacterial population and unlike wild-type T7, had no impact on the population's growth. This study shows that whilst biocontainment of phages may improve the biosafety of phage therapy, it comes at the cost of its propagation efficacy and niche penetration in relevant environments.
|
|
Scooped by
mhryu@live.com
July 3, 3:35 PM
|
Glacier forelands undergo a transition from oligotrophic to eutrophic conditions during primary succession. Reduced sulfur compounds may serve as an energy source for early microbial colonizers, yet the sulfur oxidation potential and key taxa remain largely unknown. Here, we perform a multi‑omics survey across a 130‑year chronosequence on the Tibetan Plateau. Glacial retreat profoundly reshapes both viral communities (61,394 viral operational taxonomic units, vOTUs) and microbial communities (404 metagenome‑assembled genomes, MAGs). Notably, Oxidative Dissimilatory sulfite reductase (Dsr) operon‑encoding Sulfur‑Oxidizing Bacteria (ODSOB) were specifically enriched within the first 1–5 years after retreat. Their associated viruses predominantly follow a “piggyback‑the‑winner” strategy, influencing host cold shock protein evolution and potentially modulating sulfur oxidation via iron‑sulfur (Fe‑S) cluster assembly. Metatranscriptomics reveals elevated expression of the oxidative Dsr operon and Form‑I ribulose‑1,5‑bisphosphate carboxylase/oxygenase (RubisCO) in early stages, coinciding with higher sulfate, sulfite, sulfide, and dissolved inorganic carbon (DIC)‑to‑dissolved carbon ratios compared to later stages. These findings indicate that ODSOB support DIC fixation and sulfide detoxification during early ecosystem development. Collectively, this study uncovers the eco‑evolutionary dynamics between viruses and microbes in developing ecosystems and provides genomic and functional evidence for ODSOB as key drivers of soil formation and primary succession in glacial forelands. This study shows that in glacier forelands sulfur-oxidizing bacteria and associated viruses are enriched within 1–5 years after glacial retreat. These microbes alter carbon fixation and sulfide detoxification, acting as drivers of primary succession.
|
|
Scooped by
mhryu@live.com
July 3, 3:21 PM
|
Global livestock production faces mounting pressures from climate variability, heat stress, emerging diseases, antimicrobial resistance, and sustainability demands, necessitating transformative nutritional strategies. Next-generation approaches highlight postbiotics, functional amino acids, microbial proteins, nanotechnology-enabled delivery, and nutrition-driven epigenetic regulation. Postbiotics, including short-chain fatty acids and peptides, enhance gut integrity, immunity, and feed efficiency while reducing antibiotic reliance. Functional amino acids modulate the NRF2–KEAP1 pathway, facilitating hepatic proteome remodeling and stress resilience. Sustainable alternatives, such as microbial proteins and nanocarriers, improve nutrient bioavailability with lower environmental costs. Nutritional epigenetics offers a paradigm shift, as one-carbon metabolites (methionine, choline, folate, vitamin B12) influence DNA methylation, histone dynamics, and microRNA regulation, shaping growth, reproduction, and thermotolerance. Future innovations include epigenome-guided diets, parental nutritional programming, digital twin models, and synthetic biology-driven bioactives. Collectively, these advances redefine nutrition as a driver of resilience and sustainability, requiring interdisciplinary integration, rigorous validation, and equitable translation into practice.
|
|
Scooped by
mhryu@live.com
July 3, 2:40 PM
|
Energy-conserving mechanisms are essential in supporting cellular life. Yet in synthetic biology, it remains a challenge to reconstruct such processes from the bottom–up and integrate them with other biological functions to create complex systems with life-like properties. Recent efforts to build higher-order cell-free metabolic networks have suffered from the fact that their central oxidation reactions are not coupled to energy conservation, causing kinetic and thermodynamic limitations. Here, we developed an artificial respiratory chain that we tailored to sustain rapid electron transfer in a CO2-fixing 16-enzyme catalytic cycle (crotonyl-CoA/ethylmalonyl-CoA/hydroxybutyryl-CoA), while also exploiting the concurrent electron flow for adenosine triphosphate synthesis. We demonstrate how such artificial respiratory chains can be further diversified to accept multiple electron entries and coupled to other biological functionalities, such as cell-free transcription–translation networks. Altogether, our work highlights the opportunities and challenges of directly integrating energy conservation mechanisms when building toward self-sustaining/self-energizing artificial life-like systems.
|
|
Scooped by
mhryu@live.com
July 2, 11:56 PM
|
Virological research has traditionally focused on individual viruses or viral families. Advances in DNA synthesis now allow large-scale construction of individual gene products, enabling systematic exploration of the virome. Here, we developed a barcoded library of ∼12,000 viral open reading frames (vORFs) from 513 viral species, which we leveraged to identify hundreds of viral regulators of cellular proliferation, MHC class I antigen presentation, and interferon signaling. Integrating results across these screens revealed unique phenotypic profiles and functional vORF modules, allowing the in-depth characterization of two previously uncharacterized viral proteins, MC162R and Yaba-like disease virus (YLDV) 151R, which impair MHC class I antigen presentation and interferon (IFN)-β signaling, respectively. Together, the viral ORFeome provides a scalable framework for dissecting viral protein function across the breadth of the virome.
|
|
Scooped by
mhryu@live.com
July 2, 11:37 PM
|
Plants operate as metaorganisms, depending on the coordinated signalling between the microbiomes of the roots (rhizosphere) and leaves (phyllosphere). This review covers recent studies that have identified rhizosphere-phyllosphere cross-talk as a crucial determinant of systemic stress resilience. Microbial metabolites, phytohormones, volatile organic compounds (VOCs), extracellular vesicles (EVs), and short RNAs (sRNAs) coordinate subterranean responses via vascular, gaseous, and molecular routes. Beneficial root-associated microbes modulate plant ethylene levels and antioxidant defense system in leaves through production of indole-3-acetic acid (IAA) and ACC deaminase activity. This causes the leaves to hold more water and chlorophyll when it is dry. In contrast, phyllosphere methylotrophs control root exudation through cytokinin-linked feedback which maintains metabolic balance. The identification of EV-encapsulated sRNAs and microbial lipopeptides as mobile nano-messengers paves way for a novel epoch in plant-microbe communication. Fungi, mycorrhizal association, and polyphagous insects are important in the regulation of nutrient fluxes and mediation of the trade-offs between nutrient acquisition and plant defense. Integrative multi-omics, isotope tracking, and synthetic community (SynCom) reconstructions now enable causal mapping of these systemic linkages. Understanding the cross-talk between different parts of the microbiome can help develop climate-resilient crops and provide a mechanistic basis for sustainable agriculture.
|
|
Scooped by
mhryu@live.com
July 2, 11:32 PM
|
DNA replication is initiated at specific chromosomal loci termed origins. In bacteria, the master replication initiation protein DnaA unwinds the origin (oriC), allowing a pair of replicative helicases to be loaded around each strand of the DNA duplex. The molecular mechanisms for managing bacterial helicase loading at oriC are unclear. Here we have investigated the role of the essential accessory helicase loader DnaB in Bacillus subtilis. By identifying and characterizing DnaB residues that are critical for its role during DNA replication initiation, we have located three necessary protein–protein interactions that DnaB makes with initiation proteins DnaA, DnaD, and DnaI. Combining single particle cryo-electron microscopy, AlphaFold3 predictions, and two-hybrid interaction analyses, the data suggests that DnaB acts as an interaction hub to orchestrate dual helicase loading at the origin. We propose a model for DNA replication initiation in B. subtilis and related Firmicutes pathogens that employ DnaB-type helicase loaders.
|
|
Scooped by
mhryu@live.com
July 2, 11:12 PM
|
Polymerase-mediated DNA synthesis is fundamental to numerous biotechnology applications, but existing programmable synthesis methods depend on exchanging DNA building blocks, thereby increasing reagent use and complicating multistep workflows. Here, we introduce the TEmperature Mediated Primer Exchange Reaction (TEMPER), a programmable platform for arbitrary DNA synthesis that operates solely through temperature control without solution exchange. TEMPER uses hairpin DNA as temperature-responsive building blocks that define specific temperature range for DNA synthesis. The temperature range is determined by the length design of the hairpin, which regulates thermodynamic interactions between DNA molecules and allows selective and sequential DNA synthesis in one-pot. We validate its versatility by developing a DNA data storage writer, a colorimetric temperature indicator, and a temperature data logger, highlighting its broad potential in nanotechnology and biotechnology applications. Polymerase-mediated DNA synthesis has numerous potential biotechnological applications. Here the authors develop TEmperature Mediated Primer Exchange Reaction (TEMPER), a programmable one-pot DNA synthesis method that stores data in DNA via temperature cycles and records thermal history.
|




























Using transgenic Arabidopsis expressing the Ca2+ biosensor GCaMP331, we observed [Ca2+]cyt changes in intact plants following exposure to VOCs emitted by damaged plants in real time.