Your new post is loading...

|
Scooped by
?
Today, 1:27 AM
|
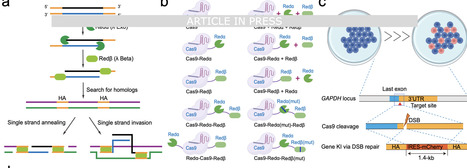
CRISPR-Cas9 tools have revolutionized genetic engineering, yet the efficient precise integration of DNA cargos, particularly for large DNA payloads (>1 kilobase, kb), remains a technical bottleneck. Herein, we develop a Recombinases (Redα/β)-enhanced DNA integration-CRISPR-Cas9 approach, referred to as RED-CRISPR, which offers a versatile yet robust homology-directed repair (HDR) strategy enabling efficient and precise kb-scale DNA insertion across various cell types, including immortalized and primary cells of variable origins. RED-CRISPR significantly enhances HDR efficiencies by 2- to 5-fold change across diverse loci and further elevates HDR rates by 1.5- to 2.5-fold when synergizing with other HDR-enhancing strategies. We achieved up to 45% knock-in efficiency for CAR-T cell manufacturing, and attained 43% knock-in rate for generation of genetically modified mice using an 8-kb DNA cargo. Through a head-to-head comparison, RED-CRISPR profoundly mitigates off-target mutational burden and chromosomal translocations. We envision RED-CRISPR as a powerful genome-editing tool with broad biomedical and therapeutic applications. Insertion of a long DNA sequence into the host genome is challenging in mammalian cells. Here, the authors develop a recombinase (Redα/β)-enhanced DNA integration approach, which enables efficient and precise kilobase-scale DNA insertion in both primary cells and mouse embryos.
|
|
Scooped by
?
Today, 12:03 AM
|
Human immune protection against bacteria critically depends on activation of the complement system. The direct bacteriolytic activity of complement molecules against Gram-negative bacteria acts via the formation of Membrane Attack Complex (MAC) pores. Bactericidal MAC pores damage the bacterial outer membrane, leading to destabilization of the inner membrane. Although it is well-established that inner membrane damage is crucial for bacterial cell death, the critical event causing MAC-mediated inner membrane damage remains elusive. Here we question whether the bacterial cell envelope possesses vulnerable spots for MAC pores to insert. By following the localization of MAC pores on E. coli over time using fluorescence microscopy, we elucidate that MAC deposition initiates at the new bacterial pole, which induces inner membrane damage and halts bacterial division. MAC components C8 and C9 preferentially localize at new bacterial poles, while C3b localizes randomly on the bacterial surface. This suggests that preferential MAC localization is determined by one of the initial steps of MAC formation. These findings provide valuable information about the interplay between immune components and the Gram-negative cell envelope.
|
|
Scooped by
?
December 9, 11:43 PM
|
Substitution matrices like BLOSUM62 model the likelihood of replacement of amino acids in evolution. Substitution matrices are used in protein sequence alignment tasks. Since the introduction of BLOSUM62 over three decades ago, many matrices have been released. Yet, to date, no effort uses large amounts of 3D structures predicted by AlphaFold. Here, we define AFSM, the AlphaFold Substitution Matrix derived from over 20,000 predicted 3D structures following the BLOSUM methodology. We benchmark AFSM against BLOSUM62 and 16 other matrices on five tasks in multiple sequence alignment (MSA) and protein homology search. Our analysis surprisingly reveals that all matrices perform similarly. Only when there are few sequences in an MSA, then BLOSUM62 and AFSM perform better than using no matrix. This suggests that substitution matrices were most beneficial when there was little sequence data. We corroborate this argument by showing that embeddings, which are computed from billions of sequences, perform better than substitution matrices, when sequence data is sparse. Taken together this suggests that structural data does not improve BLOSUM62. But increased sequence data makes extrapolation with substitution matrices obsolete. Nonetheless, BLOSUM62 continues to capture chemists intuition on amino acids by providing numerical values implicitly reflecting physicochemical properties, and it remains indispensable for direct comparison of two sequences.
|
|
Scooped by
?
December 9, 11:32 PM
|
High-yield biomanufacturing requires large cell populations and a mechanism for directing metabolic resources towards product synthesis. However, the resources that support population growth are the same as those that drive productivity, creating a conflict that limits production yields. To overcome this fundamental limitation, we apply the principle of division of labor to separate reproductive and metabolic tasks into distinct cell types within an isogenic Saccharomyces cerevisiae culture. We introduce MiSTY (Microbial Stem Cell Technology in budding Yeast), a genetic platform that exploits natural asymmetric cues to control cell differentiation. Leveraging bud cell-specific transcription, a sequential series of recombinase-based genetic circuits generates Activated Stem Cells (ASCs) that divide asymmetrically into two cell types: bud cells that terminally differentiate into Factory Cells (FCs) and mother cells that remain self-renewing ASCs. Time-lapse microscopy demonstrated 100% differentiation fidelity across 97 cell divisions. Phenotypic and genotypic analyses showed that stem cell populations could be converted to over 95% FCs within 24 generations. By converting FCs into leucine auxotrophs, we inhibited FC proliferation while allowing continued ASC division, demonstrating complete uncoupling of cell growth from product synthesis. Because they continuously generate healthy new FCs, MiSTY cultures maintain high levels of productivity even under conditions that severely impair the growth and biosynthetic capacity of metabolically exhausted factory cells.
|
|
Scooped by
?
December 9, 5:11 PM
|
Synthetic methylotrophy offers opportunities for sustainable chemical and biofuel production. While recently established methylotrophic E. coli can grow on methanol, undesirable formate accumulation occurs during growth and bioproduction. Here, we show that NAD-dependent methanol dehydrogenase Mdh2 from Cupriavidus necator inherently overoxidizes methanol to formate, a trait we find to be widespread among NAD-dependent Mdh enzymes. In contrast, Mdh/Mdh1 enzymes from Bacillus methanolicus exclusively oxidize methanol to formaldehyde without overoxidation, as we validate in vitro for Mdh Bm MGA3 with and without activator protein Act. Since only formaldehyde is assimilated via the ribulose monophosphate pathway, this explains the physiological role of Mdh/Mdh1 paralogs in natural methylotrophs and highlights the importance of selecting appropriate Mdh variants for synthetic methylotrophy. We demonstrate methanol-dependent growth using non-overoxidizing Mdh Bm MGA3, strongly reducing formate accumulation and carbon loss. Our findings reveal a characteristic of NAD-dependent Mdh enzymes and provide insights for engineering synthetic methylotrophs. Formate produced by synthetic methylotrophic E. coli can lead to carbon loss and negatively impact bioproduction efficiency. Here, the authors report the production of formate as a widespread property of NAD-dependent methanol dehydrogenases and identify Mdhs without this overoxidation activity.
|
|
Scooped by
?
December 9, 4:29 PM
|
Genome editing has revolutionized plant biology research. However, efficient and straightforward delivery of editing reagents remains a major challenge. Viral delivery systems can address these issues, but CRISPR-Cas nucleases are often too large for viral vectors. Recently, smaller editors like TnpBs have been identified, but wild-type TnpBs are significantly less active than commonly used Cas9 nucleases. Here, we optimized a tobacco rattle virus (TRV)-based system to deliver newly discovered, highly active engineered ISDra2 TnpB variants. Our results demonstrate that the eTnpBc variant delivered via TRV enables effective somatic editing in systemic leaves and achieves up to 90% editing efficiency at target loci, significantly higher than that of wild-type ISDra2 TnpB. Additionally, up to 89% of offspring exhibit a mutant phenotype, with editing efficiencies reaching 100%. The design principles outlined here are expected to accelerate broader adoption of eTnpBc for transformation- and transgene-free genome editing in plants.
|
|
Scooped by
?
December 9, 4:03 PM
|
Dietary oligosaccharides are prebiotics that fuel gut microbes, but individual microbiomes may respond differently depending on oligosaccharide structures as well as microbiome composition and function. The extent to which specific gut microbial communities exhibit personalized functional responses to distinct oligosaccharides remains underexplored. We applied a standardized ex vivo microbiome culture, called RapidAIM, coupled with metaproteomics to examine how six structurally diverse oligosaccharides affect the gut microbiota functional response. Our study shows that while human gut microbiomes share some commonalities in utilizing oligosaccharides (e.g. prioritizing dietary fibers over mucin), the fine-scale metabolic and taxonomic responses are highly individualized. Such findings underscore the importance of considering personal microbiome profiles when predicting the outcome of prebiotic interventions. In a broader context, our metaproteomic approach provides a framework for identifying optimal prebiotic choices tailored to individual microbiomes. Ultimately, understanding these personalized responses could inform precision nutrition strategies.
|
|
Scooped by
?
December 9, 3:27 PM
|
Trait-based approaches are increasingly applied to elucidate the microbial mechanisms that drive nutrient cycling and plant productivity in the rhizosphere. Genomic traits constraining trade-offs among functional traits are emerging as critical dimensions of ecological strategies. Although phenotypic traits have been studied extensively, the ecological relevance of genomic traits in shaping ecological strategies remains unclear. Here, we propose that genome size and guanine-cytosine content constitute core axes that integrate genomic architecture with fungal trade-offs in growth yield, resource acquisition, and stress tolerance. We synthesize current evidence on how genomic traits adapt to environmental gradients and how they influence fungal ecological strategies that modulate plant–fungi interactions. Advancing this conceptual framework promises deeper insight into trait–environment dynamics and plant–microbe interactions across environmental gradients.
|
|
Scooped by
?
December 9, 2:27 PM
|
The human gut virome represents a critical yet underexplored component that regulates bacterial communities and maintains gut health. However, virome analysis remains challenging due to the vast diversity and genomic variability. Existing profiling methods often struggle with accuracy and efficiency, hindering novel viral species detection and large-scale analyses. Here, we present signature-protein-based virome profiling (SinProVirP), a signature-protein-based genus-level virome profiling tool. By analyzing 275,202 phage genomes to establish a database of 109,221 signature proteins across 6,780 viral clusters (VCs), SinProVirP achieves genus-level phage quantification with accuracy comparable to the benchmark method while reducing computational demands by over 80%. Crucially, SinProVirP outperforms existing tools in detecting novel viruses, achieving over 80% recall. Applied to inflammatory bowel disease (IBD) cohorts, SinProVirP revealed disease-specific virome dysbiosis, identified high-confidence phage-host interactions, and improved the performance of bacteria-only disease classification models. SinProVirP enables robust cross-cohort virome analysis and improves our understanding of the virome’s role in health.
|
|
Scooped by
?
December 9, 1:53 PM
|
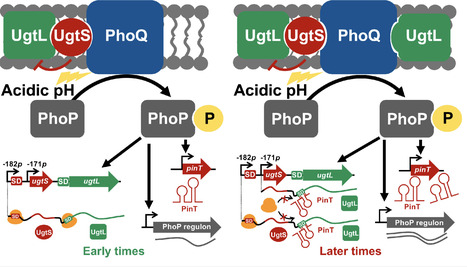
Signal transduction allows bacterial pathogens to sense the host environment and regulate gene expression accordingly for adaptation and survival. While the success of infection largely depends on the timely induction of virulence genes, the activity of the regulatory pathways controlling their expression must be tightly regulated for pathogens to cause disease. Here, we establish that a small RNA (sRNA) promotes the negative feedback control of a master virulence regulator in Salmonella enterica serovar Typhimurium by repressing a signaling protein essential for its induction in response to an intracellular cue. We show that the virulence regulatory PhoP/PhoQ pathway is inhibited by the PhoP-activated sRNA PinT in mildly acidic pH, an infection-relevant condition encountered by S. Typhimurium inside macrophages. PinT directly represses the translation of ugtL mRNA, which encodes the PhoP activator UgtL. This negative feedback regulation reduces PhoP activity, thereby decreasing the expression of PhoP-activated virulence genes like pagC. PinT-mediated repression of ugtL is predicted to be conserved in Salmonella enterica, but not in the nonpathogenic species Salmonella bongori, thus suggesting that the regulation is relevant for virulence. Our findings uncover how pathogens achieve proper levels of induction of their virulence programs through the post-transcriptional negative feedback regulation of factors enhancing the signaling activity of virulence pathways.
|
|
Scooped by
?
December 9, 1:44 PM
|
As genome-wide data have become increasingly available, software libraries for their analysis have proliferated. While new tools for downstream analyses are constantly emerging, existing workflows are hindered by inefficiencies. Switching between coding languages and object types in the early stages of pipelines wastes researchers' time, impedes reproducibility and creates opportunity for error. To confront these obstacles, we introduce tidypopgen, a comprehensive R package for population genetic analysis of biallelic single-nucleotide polymorphism (SNP) data. Genotype data can be read, filtered and analysed within a single environment, without the need for prior data cleaning or setup with other software. tidypopgen's gen_tibble object structure makes analysis efficient and intuitive, while standardized tidy grammar makes data manipulation clear. Functionality within tidypopgen supports cleaning and merging datasets, basic descriptive statistics, multivariate analysis, clustering algorithms and F-statistics, as well as integrating with existing tools for population genetic analyses in R. We use the Human Genome Diversity Project SNP dataset (Li et al., 2008) to show that a basic population genetic workflow can be executed in under 25 lines of code in a single environment using one file set, without the need to write superfluous outputs or change directories. By supporting data assembly through to data analysis, tidypopgen significantly streamlines workflows without compromising speed or functionality.
|
|
Scooped by
?
December 9, 1:23 PM
|
Next-generation therapies are advancing beyond small molecules and proteins toward engineered living microorganisms that interact symbiotically with their host and respond to signals precisely when and where needed. Despite progress in the field, engineering cells to both produce biopharmaceuticals and achieve site-specific recruitment remains a challenge. In this work, we genetically engineered the mating pathway of S. pombe to create a “bioprocessor” that responds to a chemical trigger, an artificial replica of the sexual pheromone of the yeast cells, the P-factor, enabling functional control over the production of Albulin as a proof-of-concept biopharmaceutical. This activation simultaneously induces the expression of hydrophobic agglutinins on the cell surface, modifying surface chemistry and adhesion properties. Exploiting this modification, we could simultaneously implement spatial control, allowing selective adhesion to a hydrophobic target surface. Adhesion control tests confirmed the fundamental role of hydrophobic interactions in this adhesion process, enabling selective cell adherence only after activation with P-factor and expression of the agglutinins, even in presence of potentially interfering cells. This approach represents an important milestone in the development of a straightforward chemically-activated multi-control mechanisms, which enable precise and programmable responses in engineered cells. Such advancements pave the way for a new generation of bio-responsive materials and therapeutic devices, including functional implants and targeted delivery systems, where engineered cells can operate in synergy with host tissues, responding to specific environmental cues to produce therapeutic agents exactly when and where they are needed.
|
|
Scooped by
?
December 9, 1:50 AM
|
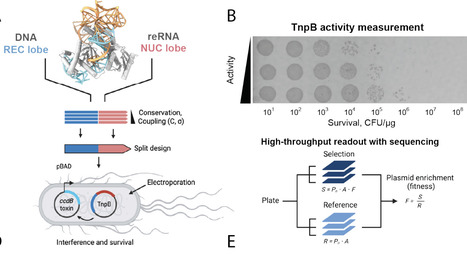
The design of RNA-guided nucleases with properties not limited by evolution can expand programmable genome editing capabilities. However, generating diverse multi-domain proteins with robust enzymatic properties remains challenging. Here we use an artificial intelligence-driven strategy that couples structure-guided inverse protein folding with evolution-informed residue constraints to generate active, divergent variants of TnpB, a minimal CRISPR-Cas12-like nuclease. High-throughput functional screening of AI-generated variants yielded editors that retained or exceeded wild-type activity in bacterial, plant and human cells. Cryo-EM-based structure determination of the most divergent active variant revealed new stabilizing contacts in the RNA/DNA interfaces across conformational states, demonstrating the design potential of this approach. Together these results establish a strategy for creating non-natural RNA-guided nucleases and conformationally active nucleic acid binders, enlarging the designable protein space.
|
|
|
Scooped by
?
Today, 1:20 AM
|
Streptomyces species are prolific producers of bioactive natural products, yet their genetic manipulation remains constrained by inefficient DNA delivery methods in many strains. Conjugation from methylation-deficient Escherichia coli has become the preferred approach for introducing plasmids into Streptomyces, relying on the presence of the oriT sequence within the mobilizable plasmid and the conjugation machinery (tra genes) encoded on the non-mobilizable helper plasmid pUZ8002. Among these, traJ encodes an essential component of the relaxosome. An additional copy of traJ is present downstream of oriT in some mobilizable plasmids, whereas many other commonly used plasmids lack traJ. Here, we investigated the impact of including traJ in mobilizable plasmids on conjugation efficiency by engineering two oriT-containing plasmids that initially lacked traJ: the ΦC31 integrative vector pRASK-SP44 and the non-replicative transposon delivery vector pHL734. We also examined the effect of introducing a second copy of traJ into the recombination-based chromosomal end-removal vector pCER. Incorporation of traJ into pRASK-SP44 and pHL734 resulted in tenfold and 100-fold increases in transconjugant numbers, respectively. Furthermore, introducing a second copy of traJ into pCER led to a fivefold improvement in plasmid transfer. Our data suggest that the inclusion of traJ improves transfer efficiency and may help overcome limiting steps in conjugation from E. coli to Streptomyces. Modulating the presence and copy number of traJ could represent a simple yet effective strategy to enhance genetic accessibility in Streptomyces. These findings have broad implications for the optimisation of genetic tools used in Streptomyces genome engineering and natural product discovery.
|
|
Scooped by
?
December 9, 11:59 PM
|
Microbial translation arrest peptides monitor intracellular environments and feedback-regulate downstream gene expression. Previous studies have identified a class of bacterial arrest peptides with C-terminal RAPP-like sequences, encoded upstream of genes involved in protein localization. In this study, we found that among RAPP-like sequences, RAPP (Arg-Ala-Pro-Pro) and RGPP (Arg-Gly-Pro-Pro) could more readily evolve into translation-impeding sequences with a particularly robust arrest that is refractory to EF-P. RAPP-like motifs were found to be strongly excluded from bacterial proteomes, likely reflecting the risk of disrupting the cellular translation system. Meanwhile, these motifs tended to occur near the C-terminus of relatively small secretory and membrane proteins. Notably, they were encoded upstream of genes with diverse functions beyond protein localization. Indeed, we identified seven RAPP/RGPP-containing arrest peptides from Streptomyces lividans encoded upstream of genes with diverse functions. These findings illustrate the bidirectional evolution of RAPP-containing proteins: their elimination from bacterial proteomes and their adaptation into arrest peptides with various regulatory roles.
|
|
Scooped by
?
December 9, 11:40 PM
|
CRISPR-Cas systems offer a viable alternative to traditional detection and diagnosis methods. However, their effectiveness relies heavily on the selection of appropriate guide RNA sequences. Existing gRNA design tools were primarily developed for gene editing and are not always directly applicable to CRISPR-based detection assays. In particular, alignment-based methods are still used to estimate gRNA specificity, even though they can miss a substantial portion of off-target sites. In this work, we introduce CRISPR-DA, a CRISPR gRNA design tool for detection assays. We show that it provides a better assessment of gRNA specificity than BLAST, which detected only 33.27 \% and 0.43 \% of cross-species off-targets in two datasets. Additionally, CRISPR-DA ran two and six times faster than BLAST on these datasets, respectively. Our method incorporates advances from gene-editing guide RNA design tools, including uncertainty-informed guide RNA design, to improve the selection of guides with high on-target activity. CRISPR-DA is available at https://github.com/bmds-lab/CRISPR-DA
|
|
Scooped by
?
December 9, 10:29 PM
|
Protein-protein interactions (PPIs) are fundamental to biological processes and central to understanding disease mechanisms, making them essential for drug discovery, peptide-based therapeutics, and vaccine development. Identifying conserved structural arrangements within PPI interfaces can provide valuable insights into molecular recognition and interaction mechanisms. Here, we introduce PRIorI (PRotein-PRotein InteractiOn gRaph Isomorphism), a web-based platform to explore precomputed protein-protein interaction networks and identify conserved atomic-level interaction motifs within structural complexes. Unlike traditional methods, PRIorI models PPI interfaces as bipartite graphs, applying graph isomorphism techniques to efficiently retrieve interaction patterns independent of sequence alignment or structural superimposition. Users can query precomputed PDB interaction graphs, upload custom protein structures, or design structural motifs for targeted searches. To illustrate its applicability, we used PRIorI to analyze the SARS-CoV-2 Spike-ACE2 interface, identifying key interaction motifs previously reported in the literature. The platform successfully identified salt bridges, hydrogen bonds, and hydrophobic interactions that stabilize the viral-host complex, demonstrating PRIorI's utility in protein interaction analysis. https://priori.ufv.br/
|
|
Scooped by
?
December 9, 4:40 PM
|
The origin of life (OoL) is a fundamental and long-standing scientific question. Although a variety of plausible hypotheses had been put forward, how life began on the prebiotic Earth from a pile of prehistoric inert chemicals (gases) is still a puzzle to us. Here, to unify the existing hypotheses to cover the entire scenarios, the author proposed the “nanozymes hypothesis” of the OoL on Earth, in which natural mineral nanozymes (MN-zymes) and their later upgraded organic/inorganic hybridized nanozymes played multiple key roles in the initial emergence of life molecules, especially in the manner of “inorganic photosynthesis” under primitive Earth conditions. Under the hypothesis framework, proteins, DNA, and RNA might emerged near-simultaneously, as a result of the diversity of nanozymes and catalyses, and multiple physical and chemical key roles of the MN-zymes. Besides nanozyme aspects, several fundamental and key issues on the topic are briefly discussed and several essential elements and conditions for the natural selection and survival of life molecules are proposed.
|
|
Scooped by
?
December 9, 4:25 PM
|
This study characterizes Penicillium melinii, an endophytic fungus isolated from Arabidopsis thaliana roots, as a plant growth-promoting fungus with potential use as a model to study root development and as a biostimulant for sustainable agriculture. Although endophytes are known to promote plant growth, the underlying molecular mechanisms often remain poorly understood. Here, we aimed to elucidate how P. melinii enhances root system development and to assess its applicability across different crops. Phenotypic assays were conducted in Arabidopsis, quinoa and tomato under in vitro, greenhouse and field conditions. Root architecture and biomass were quantified using image-based phenotyping. Transcriptomic and phytohormone profiling assessed plant responses, and fungal genome sequencing coupled with secretome analysis was used to identify candidate effectors and metabolic traits. P. melinii consistently promoted root growth and increased plant biomass across species and environments, both in vitro and in the greenhouse. In tomato field trials, this translated into a significant increase in yield. The fungus colonized root surfaces without vascular penetration and triggered a mild transcriptomic response: early activation of stress-response genes followed by their attenuation and sustained upregulation of auxin-related pathways. Notably, the interaction modulates the SLR-ARF-LBD pathway and the number of pre-branch sites probably through increased auxin signalling in the oscillation zone. Additional hormonal changes were limited and mainly associated with the attenuation of the plant response to microorganisms. P. melinii enhances lateral root formation through a subtle molecular and metabolic dialogue with the host plant, underscoring its relevance as a model for studying root developmental plasticity. Its strong and reproducible growth-promoting effect, demonstrated with different fungal strains and under controlled and field conditions, supports its potential as a biostimulant for sustainable crop production.
|
|
Scooped by
?
December 9, 3:51 PM
|
In Gram-negative bacteria, the enzymatic modification of Lipid A with aminoarabinose (L-Ara4N) leads to resistance against polymyxin antibiotics and cationic antimicrobial peptides. ArnC, an integral membrane glycosyltransferase, attaches a formylated form of aminoarabinose to the lipid undecaprenyl phosphate, enabling its association with the bacterial inner membrane. Here, we present cryo-electron microscopy structures of ArnC from S. enterica in apo and nucleotide-bound conformations. These structures reveal a conformational transition that takes place upon binding of the partial donor substrate. Using coarse-grained and atomistic simulations, we provide insights into substrate coordination before and during catalysis, and we propose a catalytic mechanism that may operate on all similar metal-dependent polyprenyl phosphate glycosyltransferases. The reported structures provide a new target for drug design aiming to combat polymyxin resistance. The study reveals the structure of ArnC, a membrane glycosyltransferase that modifies undecaprenyl phosphate with aminoarabinose (L-Ara4N), leading to polymyxin resistance. The structure of ArnC in two states and MD simulations provide insights regarding the catalytic cycle of the enzyme.
|
|
Scooped by
?
December 9, 2:38 PM
|
Photosynthesis, vital for life on Earth, is influenced by atmospheric CO2. Lu et al. recently found that introducing a novel malyl-CoA glycerate (McG) cycle into Arabidopsis thaliana works with the native Calvin–Benson–Bassham (CBB) cycle, and boosts growth, lipids, and seed yield by bypassing photorespiration, offering new strategies for crop improvement under ambient CO2 levels.
|
|
Scooped by
?
December 9, 2:14 PM
|
Within legume root nodules, rhizobia differentiate into bacteroids, which reduce N2 into NH3 for secretion to the plant. Bacteroids may be swollen and terminally differentiated or non-swollen and can regenerate outside nodules. It is unclear why these different endosymbiotic lifestyles exist and whether they differ in symbiotic efficiency. Here, we compared N2 fixing bacteroids of the near isogenic strains Rhizobium leguminosarum bv. phaseoli 4292 (Rlp4292) and R. leguminosarum bv. viciae A34 (RlvA34), nodulating Phaseolus vulgaris (common bean) and Pisum sativum (pea), respectively. The larger bean plants fixed more N2, but peas fixed 1.6-3-fold more per unit nodule mass. Values per unit volume were similar between bean and pea because bean nodules are 2.7-fold denser (i.e., mass per unit volume). Bean nodules have higher numbers of smaller (∼1/5 the volume) bacteroids than peas. Bean bacteroids are denser (i.e., 2.5-fold protein per unit volume) although less closely packed than pea bacteroids (i.e. more space between bean bacteroids). Critically, pea bacteroids, fix N2 at higher rates versus bean per unit bacteroid protein, as protein expression is skewed towards N2 fixation and TCA-cycle enzymes. Pea bacteroids infect 1.6 times the percentage of nodule volume of beans (i.e., 14.2% versus 9.1%). Overall, the increased packing density of pea bacteroids, as well as the bias of their proteome to nitrogenase, associated N2 fixation processes, and dicarboxylate metabolism, contributes to their greater symbiotic efficiency, which is likely driven by plant antimicrobial peptides.
|
|
Scooped by
?
December 9, 1:46 PM
|
Minerals are fundamental yet underappreciated drivers of microbial ecology. Traditionally viewed as passive nutrient sources or inert scaffolds, their broader ecological roles remain poorly defined. This study investigates the evolutionary influence of substrates (minerals and rocks) on soil bacterial communities through serial passage evolution experiments. Soil-derived microbial consortia from three distinct locations were exposed to nutritive (olivine, granite, diorite) and non-nutritive (quartz, kaolinite, montmorillonite) substrates under nutrient-rich conditions to isolate substrate-specific effects. Results revealed systemic variations of community structure across all treatments, characterized by elevated Firmicutes/Bacteroidetes ratio and taxonomic changes predominantly driven by rare taxa. These discoveries indicate that, under the influence of substrates, the communities shifted toward ones that preferentially utilize more labile carbon. Crucially, the acute responsiveness of rare taxa to mineral-induced environmental selection suggests that, although abundant taxa appeared to maintain core community functions, the rare biosphere facilitated niche specialization and functional diversification. These findings position minerals as dynamic drivers of microbial ecology and evolution, highlighting the mineralosphere as a critical microhabitat where abiotic properties govern biodiversity, functional redundancy, and evolutionary innovation in soil ecosystems.
|
|
Scooped by
?
December 9, 1:32 PM
|
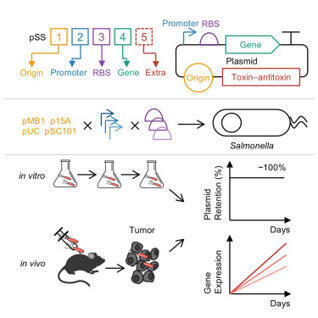
Salmonella typhimurium VNP20009 is a promising tumor-targeted vector for gene therapy, but conventional plasmid systems rely on antibiotic resistance markers and are prone to plasmid loss. To address this, we engineered and compared standardized plasmid expression vectors with different replicons, transcriptional controls, and stabilization modules in VNP20009. Among four replicons tested, pMB1 provided the best balance of stable expression and minimal effects on bacterial physiology. The incorporation of tunable promoter-RBS combinations enabled over 100-fold dynamic control of gene expression. Plasmid stabilization with the axe/txe toxin-antitoxin system maintained plasmid retention and protein expression without impairing growth. In murine tumor models (LLC and B16F10), the pMB1-AT system sustained therapeutic gene expression and enhanced tumor suppression when delivering Endostatin. This work establishes an antibiotic-free and durable plasmid platform for Salmonella-based therapies, offering a framework for reliable gene delivery in vivo and expanding opportunities for synthetic biology and cancer treatment.
|
|
Scooped by
?
December 9, 1:09 PM
|
The complexities of microbial detection and conventional enumeration necessitates the adoption of pragmatic alternatives. This review expands the boundaries of knowledge for microbial detection and sensing, particularly within the field of water quality analysis. Observed alternatives to conventional techniques for microbial analyses in recent studies include Microarray, Fluorescent in situ hybridization (FISH), loop-mediated isothermal amplification (LAMP), matrix-assisted laser desorption ionization-time of flight (MALDI-TOF) and flow cytometry, while nanosensors stood out as an alternative for microbial detection in real-time. This study presents the limitation of conventional methods of detection in water and presents nanoparticles as a detection agent with possibility of incorporation into point-of-use detection. It is notable that nanosensors are currently emerging in the detection of bacteria, viruses and other pathogens in water and have been used in the detection of bacterial pathogens than viral. Nanosensors are established as good choice for rapid water analysis with application in point-of-use and analytical devices. In the use of nanozymes, the choice over natural enzymes can be linked to their unique and excellent catalytic activities, cost-effectiveness and ease of mass production.
|