 Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 1:34 AM
|
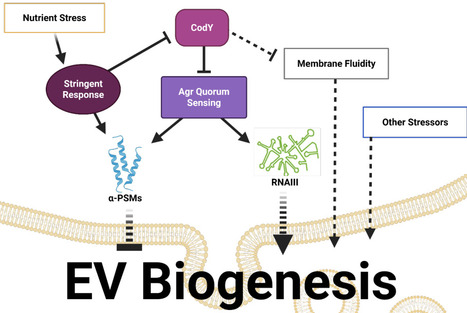
The release of extracellular vesicles (EV) is a universally conserved process. Bacterial EVs package diverse cargo, including proteins and nucleic acids, and influence bacterial adaptation and survival as well as host-pathogen interactions. Currently, our understanding of the mechanisms underlying global principles in Gram-positive EV biogenesis and release is limited, partly due to labor-intensive vesicle isolation and assessment methods. Here, we describe a moderately high-throughput approach to analyze the Nebraska Transposon Mutant Library to identify genetic determinants of EV production in S. aureus. We show that the agr quorum sensing system dictates EV production in response to nutrient availability, likely through communication with the adaptive stress response. This study demonstrates the contribution of nutritional stress to vesiculogenesis and supports a conserved communication strategy that allows metabolic state to influence EV production. omv
|
|
Scooped by
mhryu@live.com
Today, 1:21 AM
|
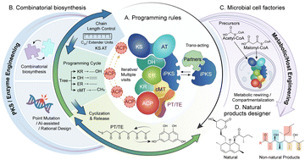
Polyketides constitute a vast family of structurally and functionally diverse natural products that underpin numerous pharmaceuticals, nutraceuticals, and materials. Among them, fungal type I iterative polyketide synthases (iPKSs) orchestrate highly programmable catalytic cycles that transform simple acyl-CoA precursors into architecturally complex molecules. Understanding the programming logic of these multidomain enzymes has revealed how chain-extension, reduction, and cyclization patterns are encoded, offering a foundation for rational pathway engineering. Recent advances in structural biology, cryo-electron microscopy (cryo-EM) analysis, and computational modelling have clarified the conformational dynamics of iPKSs and their collaborating enzymes, while combinatorial biosynthetic strategies now enable the creation of non-natural scaffolds and expanded chemical diversity. Parallel progress in fungal and yeast cell-factory engineering—spanning metabolic rewiring, organelle compartmentalization, and dynamic control—has substantially improved the efficiency and scalability of polyketide production. This review integrates mechanistic insights with biotechnological innovation, highlighting emerging rules for programmable PKS design and discussing future directions toward AI-assisted, high-throughput platforms for sustainable industrial biosynthesis of fungal polyketides.
|
|
Scooped by
mhryu@live.com
Today, 1:16 AM
|
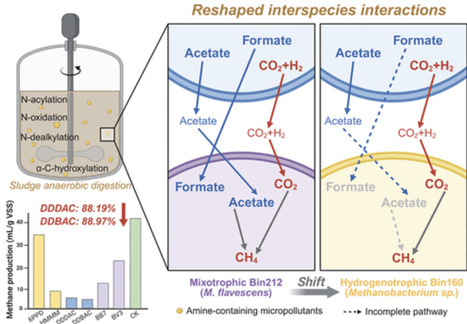
Amine-containing micropollutants (AMPs), a class of structurally diverse polar compounds characterized by one or more amine functional groups, are frequently detected in wastewater sludge. However, the anaerobic transformation of these compounds and their impacts on microbial metabolism during anaerobic digestion (AD) remain poorly understood. In this work, six representative AMPs were selected to cover 16 structurally diverse primary, secondary, tertiary amine, and quaternary ammonium functionalities. α-C hydroxylation and N-acetylation were identified as the dominant initial reactions among the detected transformation products (TP), collectively accounting for 42.6% of all identified TPs. Furthermore, compound-specific differences in metabolic disturbance were observed. Quaternary ammonium compounds, N-dodecyl-N-benzyl-N,N-dimethylammonium chloride (DDBAC) and N,N-Didodecyl-N,N-dimethylammonium chloride (DDDAC) markedly reduced acetate kinase activity by 10.69 and 14.28%, respectively, and resulted in methane production yield reductions of 88.97 and 88.19%. The genome-centric metagenome revealed that exposure to AMPs prompted the reassembly of the microbial community, altered its functional attributes, and disturbed interspecies cross-feeding interactions. Specifically, AMPs triggered a shift in the methanogenic consortium from mixotrophic Methanosarcina flavescens to hydrogenotrophic Methanobacterium sp., owing to the latter’s metabolic versatility, vigorous proliferation, and superior energy conservation. These findings indicated that the chemical properties of amine functional groups have effects on anaerobic biotransformation pathways and microbial energy metabolism, providing mechanistic insight into AMPs toxicity and guiding mitigation strategies to enhance the stability and resilience of full-scale AD systems.
|
|
Scooped by
mhryu@live.com
Today, 12:58 AM
|
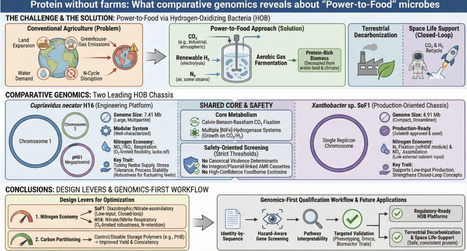
Conventional agriculture is increasingly incompatible with planetary boundaries, such as land and water demand, greenhouse-gas emissions, and disruption of the nitrogen cycle. Hydrogen-oxidizing bacteria (HOB) enable a scalable ″power-to-food″ approach in which aerobic gas fer-mentation turns CO2 and renewable H2, along with N2 in some strains, into protein-rich biomass, largely decoupling protein production from arable land and climate variability. The same chemistry is attractive for closed-loop space life support, where crew CO2 and electrolysis-derived H2 can be recycled into edible biomass. Here, we compare two leading HOB chassis strains, Cupriavidus necator H16 and Xanthobacter sp. SoF1, using standardized re-annotation, orthology-based comparison, pathway reconstruction, and safety-oriented genome screening. Importantly, SoF1 is the production strain for Solar Foods′ Solein®, a dried microbial biomass ingredient, which is approved as a novel food in Singapore and has a self-affirmed GRAS status in the United States. H16 has a larger, multipartite genome of 7.41 Mb split across two chromosomes and the pHG1 megaplasmid, whereas SoF1 is more compact at 4.91 Mb and encoded on a single replicon. Both encode Calvin-Benson-Bassham CO2 fixation and multiple [NiFe]-hydrogenase systems supporting growth on CO2/H2, but nitrogen economy differentiates the hosts. SoF1 encodes a complete nitrogen-fixation module (nifHDK) and nitrate-assimilation genes, whereas H16 lacks nif and instead encodes nitrate/nitrite respiration for oxygen-limited flexibility. Safety screening revealed no evidence of canonical virulence determinants, integron or plasmid-linked antimicrobial resistant (AMR) cassettes, or high-confidence foodborne exotoxins under strict thresholds. These results convert genome-level features into actionable design constraints for selecting and engineering food-grade HOB, strengthening robust air-to-protein bioprocesses on Earth and informing a blueprint for closed-loop, space-compatible protein production.
|
|
Scooped by
mhryu@live.com
Today, 12:38 AM
|
Mathematical models are increasingly a part of microbiological research. Here, we share our perspective on how modelling advances the discipline by: (i) enforcing logical consistency, (ii) enabling quantitative prediction, (iii) extracting hidden parameters from data, and (iv) generating intuitive understanding. We map a spectrum of modelling frameworks, from whole-cell simulations to minimal logistic growth equations, and provide interactive examples for some common frameworks. Building on this overview, we outline pragmatic criteria for choosing an appropriate level of description to capture phenomena of interest. Finally, we present a case study in modelling of microbial ecosystems from our own work to illustrate how mechanistic modelling can yield generalizable intuition. This perspective aims to be an introductory roadmap for integrating mathematical modelling into experimental microbiology.
|
|
Scooped by
mhryu@live.com
Today, 12:29 AM
|
Prime editing (PE) enables precise genetic modifications using canonical prime editing guide RNA (pegRNA), with the reverse transcription template and primer binding site (RTT–PBS) attached to the 3′ ends of CRISPR–Cas guide RNAs. Although PE ribonucleoprotein (RNP) delivery holds great therapeutic potential, its weak genomic editing capability limits therapeutic applications. Here we present structure-guided engineering of the PE complex using non-canonical pegRNAs (npegRNAs), with the RTT–PBS integrated within the single guide RNA loops, to improve PE efficiency. This approach demonstrates enhanced precise editing rates across various genomic sites and cell types, and improves therapeutic gene correction in a tyrosinaemia mouse model. Cas9-associated npegRNAs are more resistant to exonuclease degradation, probably enhancing the PE complex’s targeting efficiency in living cells. Using PE RNP delivery, npegRNAs achieve increased average editing yields of 26.8-fold over canonical pegRNAs and 5.9-fold over engineered pegRNAs (epegRNAs). Furthermore, npegRNA-mediated RNPs increased the efficiency of installing disease-relevant mutations up to 123-fold in human cell lines, including Jurkat T cells and induced pluripotent stem cells. Collectively, our findings demonstrate a robust PE strategy and highlight the potential of npegRNAs for therapeutic PE applications. Prime editing using non-canonical prime editing guide RNAs, where the reverse transcription template and primer binding site is integrated within the single guide RNA loops rather than attached to the 3′ ends of CRISPR–Cas guide RNAs, increases editing efficiency.
|
|
Scooped by
mhryu@live.com
Today, 12:11 AM
|
The NAD+ cap has been discovered in RNAs across prokaryotes and eukaryotes, suggesting a possible role of NAD capping in gene regulation. Current NAD-capped RNA (NAD-RNA) profiling methods lack precision in 5’-end mapping or bias against small NAD-RNAs. Here, we introduce precision NAD-RNA sequencing (pNAD-seq), which combines a two-step enrichment strategy with high-throughput sequencing to achieve single-nucleotide resolution of 5’-ends and unprecedented sensitivity for identifying small NAD-RNAs. We further develop NAD-linkSeq to determine full-length NAD-RNA sequences. Applying these methods to E. coli, we uncover a vast repertoire of NAD-RNAs, including tRNAs, rRNAs, intragenic transcripts, and antisense RNAs, many of which are significantly shorter than regular mRNAs, implying specialized biogenesis. High-resolution mapping reveals conserved promoter architectures driving NAD-RNA production and condition-dependent initiation dynamics: under nitrogen limitation, some NAD-RNA undergo TSS switching, alternative promoter usage, and coordinated expression shifts, correlating with metabolic stress responses. This report presents findings that offer a comprehensive view of NAD-RNAs in E. coli and introduces reliable methods for genome-wide profiling of NAD-RNAs across different organisms, which will facilitate the functional characterization of NAD-capping. Some RNAs carry an NAD+ cap that may regulate genes, but current methods miss small RNAs and precise start sites. Here, the authors develop two profiling methods that map NAD-capped RNAs at single-base resolution, uncover many new species in E. coli, and reveal stress-responsive promoter activity.
|
|
Scooped by
mhryu@live.com
April 7, 11:01 PM
|
In the post-antibiotic era, the Bacillus-based direct-fed beneficial microorganisms are emerging as a cornerstone for sustainable animal farming. This study aimed to screen and evaluate Bacillus strains with probiotic potential for use as feed additives. A total of 394 Bacillus strains were initially screened based on their extracellular enzyme production (cellulase, protease, and amylase) and antibacterial activities against E. coli, Staphylococcus aureus, and Salmonella enterica. Two strains, Bacillus velezensis FJAT-10508 and FJAT-13563, were selected and subsequently subjected to in vitro probiotic characterization, safety assessment, and whole-genome analysis. The results demonstrated that both strains exhibited α-hemolysis, acceptable antibiotic susceptibility profiles, absence of invasion and cytotoxicity effect on the Caco-2 cells, and no mobile virulence or antibiotic resistance genes, indicating their safety as probiotic candidates. High endospore-forming efficiencies (72.4–90.8%), strong auto-aggregation (74–85%) and co-aggregation abilities (52–82%) were observed. In addition, both strains showed considerable tolerance to simulated gastrointestinal conditions, with vegetative cell and endospore survival rates of 28.33–38.33% and 85–89.67% at pH 2.0, and 38.33–43.33% and 90.33–96.33% in 0.3% bile salts, respectively. Overall, B. velezensis FJAT-10508 and FJAT-13563 demonstrated robust in vitro probiotic properties, supporting their potential application as reliable Bacillus-based feed additives.
|
|
Scooped by
mhryu@live.com
April 7, 1:51 AM
|
Survival of bacteria in dense and competitive microbial communities depends on their ability to access nutrients or outcompete neighboring cells. One of the key players in competition is the type VI secretion system (T6SS), a contractile nanomachine that injects a broad variety of effectors into competitors. However, target cells do not remain defenseless. Given the lethality of T6SS-mediated attacks, they have evolved countermeasures to detect, withstand, or resist such assaults: immunity proteins that neutralize effectors, activation of envelope stress pathways to mitigate damage, retaliatory responses to avoid recurrent attacks, physical barriers such as matrix polymers, or collective protection through aggregation. In this review article, we provide a comprehensive overview of these diverse strategies and emphasize the arms-race dynamics.
|
|
Scooped by
mhryu@live.com
April 7, 12:18 AM
|
Mangroves are ecosystems located at land–sea transition zones, where they are continuously exposed to plant biomass and plastic pollution. Their soils harbor extensive microbial diversity with potential for discovering polymer-degrading enzymes. Here, we perform a microcosm experiment to examine how mangrove soil microbial communities respond to inputs of lignocellulose or polyethylene terephthalate (PET) in the presence and absence of seawater, and to explore the selection of putative PET-active enzymes (PETases) using gene- and genome-resolved metagenomics. Incubation conditions lead to a gradual increase in salinity, resulting in the enrichment of halophilic taxa, including spore-forming bacteria and archaeal species, particularly in seawater-depleted treatments. Lignocellulose input is the primary driver of soil microbial community restructuring, followed by seawater presence. In dry, lignocellulose-amended microcosms (L treatment), microbial diversity is significantly reduced, while lignocellulolytic taxa within the phyla Bacillota and Actinomycetota are enriched. Twelve potential PETases are identified in the L treatment, sharing >70% sequence similarity with known PETases, and three are predicted to be thermostable. Two putative PETases from Microbulbifer species display distinct sequence and structural features, thereby expanding the currently limited PETase sequence landscape. This study demonstrates that perturbing environmental microbiomes with plant-derived polymers represents a promising strategy for capturing novel PETases. Mangroves are increasingly exposed to plastic pollution. In this study, the authors performed microcosm experiments to show that mangrove soil microbial communities are reshaped by lignocellulose inputs yielding potential halophilic PET-degrading enzymes as promising candidates for plastic degradation.
|
|
Scooped by
mhryu@live.com
April 6, 11:00 PM
|
Large language models (LLMs) are increasingly used by plant biologists to summarize literature, generate hypotheses, and interpret experimental results. However, LLMs are unreliable sources of exhaustive, source-attributed facts, a critical limitation for the list-style queries that pervade plant biology (e.g., "list all transcription factors regulating secondary cell wall (SCW) biosynthesis in Arabidopsis"). Here, we query ChatGPT, Claude, and Gemini with such queries and demonstrate that none return complete gene lists with reliable citations. We trace these failures to how LLMs store knowledge: as statistical patterns distributed across billions of internal parameters, with no mechanism to guarantee completeness, provenance, or reproducibility. We also review fine-tuning mitigation strategies, including multi-task instruction tuning, parameter-efficient methods, and context engineering, that alleviate but do not resolve these limitations. We then discuss retrieval-augmented generation (RAG), which feeds relevant documents to the LLM at query time, and argue that while it improves source attribution, it remains impractical when answers require synthesizing information scattered across hundreds of papers. As an alternative, we advocate graph retrieval-augmented generation (GraphRAG), in which the LLM serves as a reasoning and language interface over a structured, provenance-linked knowledge graph (KG) that returns complete result sets reproducibly. We outline a practical GraphRAG architecture and survey existing plant KG resources. Finally, we discuss open challenges, including entity disambiguation, relation normalization and evidence grading, and propose a roadmap for building open, continuously updated plant KGs that can turn "read 1,000 papers" into a single reproducible query.
|
|
Scooped by
mhryu@live.com
April 6, 10:31 PM
|
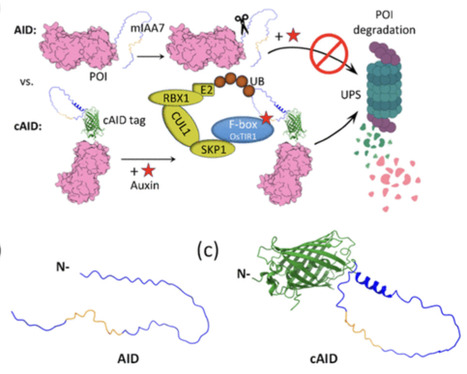
The auxin-inducible degron (AID) system enables targeted protein degradation in vivo. Conventionally, the AID tag is fused directly to the terminus of the target protein; however, its intrinsically disordered nature and destabilizing motifs can render it susceptible to premature proteolytic removal from its tagged protein, resulting in incomplete degradation upon auxin induction. In addition, direct terminal fusion of the conventional AID tag may compromise protein stability. To address these limitations, we introduce a new degron design in which the conventional AID tag, mIAA7, is inserted into an exposed loop of green fluorescent protein (GFP). The resulting engineered GFP variant, termed the “constrained AID” (cAID) tag, was validated in the industrially important oleaginous yeast Yarrowia lipolytica. Inserting mIAA7 into GFP, rather than fusing it directly at the GFP terminus, elevated expression of the GFP fusion protein and enabled more complete degradation upon auxin induction. The utility of the novel cAID tag was further validated by tagging a soluble cytosolic anti-mCherry nanobody and by targeting β-carotene ketolase, squalene synthase, and squalene epoxidase to modulate carotenoid biosynthesis. Compared to direct fusion with the conventional AID tag, cAID tag enabled more complete protein degradation and tighter temporal control of metabolism, while minimizing perturbation to the tagged protein.
|
|
Scooped by
mhryu@live.com
April 6, 4:41 PM
|
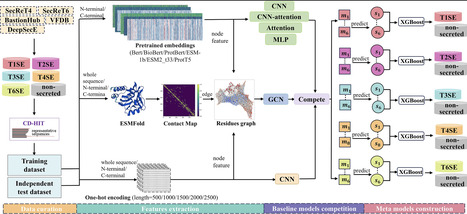
Bacterial secreted proteins, particularly effectors delivered by specialized secretion systems, are key mediators of virulence and host–pathogen interactions. However, accurate computational identification remains challenging, as many existing methods rely heavily on sequence similarity or handcrafted features, and often focus on a single secretion system. Recent studies have reported that some bacterial effectors may be associated with more than one secretion system, highlighting the complexity of secretion system annotation and motivating the development of system-aware computational prediction approaches. Here, we present PLM-Effector, a hybrid deep learning framework that integrates modern protein language models (PLMs) with multiple neural architectures via a two-layer ensemble stacking strategy. By extracting complementary features from N- and C-terminal regions, PLM-Effector enables secretion-type-aware prediction across five major bacterial secretion systems (T1SS–T4SS and T6SS), with each system modeled independently. Systematic benchmarking shows that embeddings from protein-specific PLMs (ESM-1b, ESM2_t33, ProtT5) are more discriminative than those from general-purpose language models (e.g. BERT, BioBERT). Leveraging these representations, PLM-Effector achieves superior performance on an independent test set, with macro F1-scores of 0.9848, 0.8649, 0.9899, 0.9620, and 0.9728 for secreted proteins of T1SS–T4SS and T6SS, respectively, outperforming existing tools and homology-based baselines. Implemented as an accessible web server (http://www.mgc.ac.cn/PLM-Effector/) with source code and datasets available (https://github.com/zhengdd0422/PLM-Effector/), PLM-Effector provides a reproducible and user-friendly platform for both small-scale and genome-wide secreted protein discovery, facilitating advances in the study of bacterial pathogenesis.
|
|
|
Scooped by
mhryu@live.com
Today, 1:24 AM
|
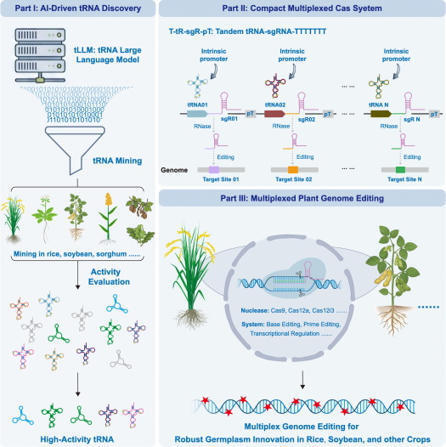
The widespread use of CRISPR-Cas9 in plants highlights the need for compact and efficient multiplexed genome editing systems. This study optimizes single-guide RNA (sgRNA) expression in CRISPR by leveraging endogenous tRNA processing mechanisms for efficient multiplexed genome editing. Screening in Arabidopsis thaliana and Oryza sativa identified superior tRNAs that outperformed the widely used AtGly-tRgcc. Leveraging tRNA’s dual functions in sgRNA processing and their intragenic RNA polymerase III promoter activity, we established a compact multiplexed system for simultaneous editing of at least ten genomic loci in rice and soybean. Moreover, we developed plant tRNA large language models that learn sequence representations to identify both canonical and noncanonical tRNAs, uncovering thousands of tRNAs missed by traditional algorithms and expanding the repertoire for genome editing. This work provides a robust tRNA-based CRISPR platform, an artificial intelligence-guided tRNA mining framework, and a comprehensive tRNA resource for advanced plant genome engineering and germplasm innovation.
|
|
Scooped by
mhryu@live.com
Today, 1:19 AM
|
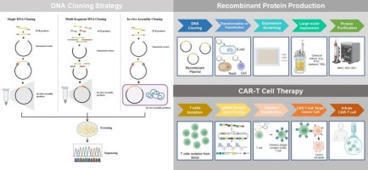
Molecular Cloning is a cornerstone technology for amplifying, expressing, and manipulating specific DNA sequences. Traditional Restriction Enzyme Cloning (REC) methods are limited by sequence dependency and the introduction of undesirable “scar” sequences, which have driven the development of more advanced assembly strategies. In this review, we critically analyze major DNA Cloning techniques—from early methods to modern seamless assembly and in vivo systems. We delineate the fundamental differences between cloning vectors, which are primarily designed for DNA amplification and stable maintenance in host cells, and expression vectors, which incorporate regulatory elements that drive the expression of recombinant proteins or the transcription of guide RNAs (gRNAs) for cell therapy applications. We describe the mechanisms and enzymes involved in each approach and evaluate their key advantages and limitations. We emphasize the distinction between “scarred” methods (e.g., Restriction Enzyme Cloning, Gateway® Cloning) and “seamless” methods (e.g., Golden Gate Assembly, Exonuclease-based Seamless Cloning), highlighting the superior precision and multi-fragment assembly capability of seamless strategies for complex DNA Cloning projects in Synthetic Biology. Finally, we compare the trade-offs between in vitro systems, which are highly efficient but costly, and in vivo assembly approaches, which are simpler and more cost-effective but typically exhibit lower efficiency—making them suitable for self-sustained academic laboratories. We conclude that the optimal cloning strategy should be selected based on the specific requirements of the project, balancing junction type (scarred/seamless), sequence dependency, multi-fragment capability, flexibility, and cost to meet the needs of each experiment.
|
|
Scooped by
mhryu@live.com
Today, 1:06 AM
|
RNase PH is a critical exoribonuclease in E. coli that participates in tRNA maturation and RNA degradation. In earlier work, it was observed that levels of RNase PH decreased as much as 90% under conditions of nutrient deprivation, such as induced starvation and prolonged stationary phase, and that its removal was likely due to the degradation of the protein. Here, we examine the mechanisms involved in this regulatory process. We find that the protease Lon is primarily responsible for the removal of RNase PH that occurs in stationary phase and starvation. Conversely, RNase PH remains stable during the exponential phase of growth due to a protective interaction with the chaperonin protein, GroEL. Overproduction of GroEL protects RNase PH even under conditions of nutrient deprivation. Additionally, we find that RNase II activity also is required for the degradation of RNase PH, implying the involvement of an RNA molecule in the overall regulatory process. In mutant strains devoid of RNase II activity, even though retaining RNase II protein, RNase PH levels remain unchanged during nutrient deprivation which leads to excessive rRNA removal and ultimately to loss of viability. These findings provide another example of the complex regulatory mechanisms that underscore the importance of maintaining appropriate RNase levels under varying physiological conditions.
|
|
Scooped by
mhryu@live.com
Today, 12:48 AM
|
Bacterial therapeutics hold great promise for cancer treatment by targeting oxygen-poor tumor regions and complementing existing therapies. However, current approaches often struggle with safety concerns and complex engineering. Developing a safe, effective delivery platform relying entirely on natural bacterial biosynthesis remains a challenge. Here we show that attenuated Serratia marcescens serves as a powerful biohybrid platform for cancer therapy by leveraging its natural biosynthesis of prodigiosin, a photosensitive pigment. We engineer S. marcescens to yield high prodigiosin levels, which exhibit strong intrinsic anti-cancer activity and near-infrared photosensitivity. In female mouse models of melanoma and colorectal cancer, this platform triggers robust systemic immune responses, including enhanced T cell recruitment and long-term memory against tumor recurrence. Furthermore, the bacteria induces tumor cell death via mitophagy, while photothermal properties of prodigiosin enables rapid, light-controlled bacterial clearance post-treatment. These findings establish S. marcescens as a versatile, self-regulating biosynthetic platform for precise and safe cancer immunotherapy. Bacterial therapies for cancer face safety and complexity challenges. Here, the authors develop an engineered Serratia marcescens platform that leverages natural pigment biosynthesis to trigger antitumor immunity and enable rapid bacterial clearance, controlled by near-infrared light.
|
|
Scooped by
mhryu@live.com
Today, 12:33 AM
|
Global food systems face challenges from population growth, shifting diets and climate change. While decades of plant-centric breeding and high-input agriculture have generated high yielding crops, this strategy has unintentionally reshaped the plant associated microbiomes, often coinciding with a depletion of their functional diversity. We revisit these breeding strategies and propose extending breeding targets beyond the plant genome to include the plant microbiome. In this regard, resistance breeding shows, albeit unintended, that plant genetics shape the microbiome: by narrowing the genetic base, we have selected for highly specialised pathogenic microbiomes. This raises a key question: can we intentionally apply the same principle to select for beneficial microbiomes? To answer this question, a thorough insight into microbial community architecture, hubs and functional redundancy is key. We outline two complementary avenues: (i) rewilding to restore ancestral microbial partners and (ii) microbiome breeding guided by QTL/GWAS mapped host loci that gate microbial recruitment, immune filtering and exudate composition. This approach comprises the integration of trait-based phenotyping, multi-omics, network-informed SynCom design and field testing across environments to capture G × E × M (genotype × environment × microbiome) interactions. Treating the microbiome as a selectable, designable and heritable trait can convert small gains into durable long-lasting crop resilience.
|
|
Scooped by
mhryu@live.com
Today, 12:17 AM
|
The microbiome is widely involved in host metabolism, with many omics studies suggesting that it is important for metabolic health. Although studies in this area have made great strides in furthering our understanding of the role of the microbiome in health and disease, key challenges still hinder the safe clinical application of gut microbiota-targeted therapies. These limitations include a lack of confirmation of causality between the gut microbiota and host health, insights into the molecular mechanisms by which the gut microbiota functions to affect host health, and the development of therapeutic strategies that accurately regulate the function of the gut microbiota towards specific microbial enzyme targets without affecting its overall composition and viability. Microbial enzymes with various functions and activities have attracted the attention of many researchers in the past few years, especially microbiota–host isozymes, which are enzymes in the microbiome and the host that share a similar function. Such isozymes, as well as microbial-specific enzymes involved in basic biological processes of the gut microbiota, metabolism of nutrients, and synthesis of active metabolites and interactions in microbial-host communities, are the key mediators of gut microbiota–host crosstalk and have received much attention. In this Review, we provide a holistic understanding of the multifaceted role of gut microbial enzymes, including providing guidance for their discovery, while highlighting the great potential of gut microbial enzyme-oriented therapies for precision medicine. In this Review, Jiang and colleagues describe how gut microbial enzymes regulate host metabolic health and diseases. They also discuss the methodology for studying gut microbial enzymes and their potential clinical applications.
|
|
Scooped by
mhryu@live.com
April 7, 11:10 PM
|
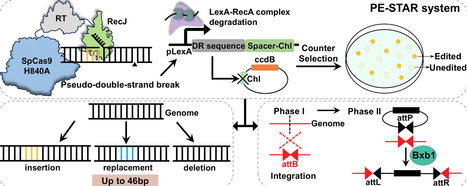
Prime editing enables precise genome modifications without DNA double-strand breaks, yet bacterial applications are limited by low efficiency and small edit sizes. Here, we develop PE-STAR, Prime Editing with SOS-Triggered and RecJ-Augmented Repair, to enhance prime editing in E. coli. Removing three inhibitory 3′→5′ exonucleases (SbcB, ExoX, and XseA) improved edited-strand retention, and extending post-transformation outgrowth increased editing efficiency. RecJ overexpression strengthened 5′-directed processing during flap resolution and gap expansion, biasing repair toward incorporation of the reverse-transcribed edited strand. To enrich edited cells, we integrated an SOS-responsive counter-selection circuit that links PE3-associated dual nicking to LexA-dependent gRNA expression targeting a plasmid encoding the toxin CcdB, thereby eliminating unedited cells. PE-STAR achieved up to 80%–90% editing efficiency for short-fragment modifications, representing up to 16-fold improvement across loci. The platform supported insertions, deletions, and replacements of up to 46 bp with high efficiency. Furthermore, installing an attB site by prime editing, followed by Bxb1 integrase recombination, enabled chromosomal integration of 3.2 and 8.0 kb cassettes with 100% recombination efficiency among screened colonies, including GFP reporter and riboflavin biosynthetic pathway. PE-STAR expands both the efficiency and functional scope of bacterial prime editing for programmable genome engineering.
|
|
Scooped by
mhryu@live.com
April 7, 1:28 PM
|
Lipid accumulation in microalgae is typically induced by stress but often comes at the expense of growth and energy stability. Here, we introduced the plsXYC module from Cyanobacterium aponinum into Chlamydomonas reinhardtii, aiming to enhance stress-responsive lipid production by a plug-in prokaryotic acyltransferase route. The initial wild-type strain produced lipids at 135.1 mg/g-dry cell, while the engineered CrXYC sustained the highest lipid yield of 385 mg/g-DCW across light and dark cycles. Transcriptomics revealed that light–dark shifts drove 65% of expression variance and elevated ATP levels. During heterotrophic culture under nitrogen starvation, CrXYC preserved ATP up to 1.6-fold higher than the parental background. Carbon repartition was proved using 13C-isotopes, and redox reinforcement showed coordinated upregulation of lipid assembly pathways and repression of starch biosynthesis. This shift coincided with enhanced superoxide scavenging activity, while broad antioxidant capacity remained unchanged. Together, we demonstrate that introducing an orthogonal acyl-acyl carrier protein entry route sustains lipid accumulation under stress while preserving growth and energy balance.
|
|
Scooped by
mhryu@live.com
April 7, 12:23 AM
|
Antimicrobial resistance (AMR) presents a pressing need to ensure that the right antimicrobials are used to target the right microbes at the right time. Ideally, the appropriate antimicrobial is selected after patient samples have been cultured and assessed with antimicrobial sensitivity testing (AST). However, the time needed for culture-based diagnosis leads to immediate empirical treatment, often with broad-spectrum and/or high-tier antimicrobials. Direct nanopore metagenomic whole genome sequencing to identify the pathogens and predict their antimicrobial resistance is a rapid and patient-side alternative. A limitation of this approach is the inconsistency of in silico predicted AMR phenotypes. Here, we benchmarked the current performance of in silico AMR prediction strategies for nanopore-generated long read data. Using nanopore data paired with AST phenotyping for 201 samples, we assessed the impact of basecalling mode, data volume, and assembly strategy to compare the performance of eight in silico AMR prediction tools with seven AMR databases. We found that basecalling accuracy mode does not affect the overall accuracy of in silico AMR predictions, but assembly strategy and data volume both do. Prediction tools using the ResFinder database scored best for balanced accuracy (0.8 for both ResFinder and ABRicate), whilst DeepARG scored best for sensitivity (0.65). However, even the best performing in silico AMR prediction strategy missed some resistance identified by lab-based AST. In silico AMR prediction can therefore supplement lab-based AST, but cannot yet replace it.
|
|
Scooped by
mhryu@live.com
April 6, 11:07 PM
|
Traditional mass spectrometry-based proteomics typically requires prior knowledge of sample composition to match spectra to peptides. Yet, novel de novo peptide sequencing approaches can provide peptide sequences to identify the organism. Here, we introduce an end-to-end pipeline (NovoTax) to identify the closest prokaryotic genome directly from raw bottom-up proteomics data. The approach combines peptide sequencing tools with an optimized implementation of peptide searching through an extensive genome database. On a benchmark dataset of species isolates, we identified the reported species and strain in the majority of the cases, and showed that in discordant cases NovoTax was likely correct. Interestingly, NovoTax was also able to identify contaminating species in some samples. The algorithm also identified the most abundant species in bacterial communities. In summary, NovoTax provides strain level identification of microbial samples enabling the downstream use of traditional proteomics search engines for a deeper proteome analysis.
|
|
Scooped by
mhryu@live.com
April 6, 10:55 PM
|
Understanding the role of gene expression in cellular function and tissue organization requires spatial and quantitative detection of individual RNA molecules. Yet, the widespread adoption of automated single-molecule fluorescence in situ hybridization (smFISH) has been limited by the cost of equipment and the complexity of experimental procedures. We present autoFISH, an affordable, user-friendly platform that removes these barriers through open-source hardware components, accessible control software, and integrated analysis tools. The system demonstrates broad applicability by enabling both conventional and signal-amplified smFISH protocols and incorporates an optimized tissue-clearing method that preserves nuclear structures. Testing across multiple cell types and tissue preparations validates the system’s reliability and reproducibility, offering a practical solution for scaling spatial transcriptomics research and advancing discoveries in cellular and developmental biology, while significantly reducing costs and the technical expertise required. AutoFISH provides a cost-effective, open-source toolbox for automated smFISH, offering integrated hardware, software, and optimized protocols to streamline spatial gene-expression profiling in cell lines and cleared tissue samples.
|
|
Scooped by
mhryu@live.com
April 6, 4:54 PM
|
A systematic understanding of cellular metabolism is essential for engineering yeast and uncovering the principles of metabolic robustness and evolution, yet much of its metabolic space remains unexplored. Although yeast genome-scale metabolic models have been reconstructed and curated for over two decades, more than 90% of the yeast metabolome remains uncovered. Here, to address this gap, we have developed an integrated workflow that combines retrobiosynthesis, deep learning-based enzyme annotation and enzyme–substrate prediction to systematically explore yeast underground metabolism. Using the framework, we reconstruct a yeast metabolic twin model, Yeast-MetaTwin, comprising 16,244 metabolites, 1,976 metabolic genes and 59,865 reactions. The model reveals systematic differences in Km distributions between the known and underground networks and identifies key hub metabolites linking the underground network. Moreover, Yeast-MetaTwin predicts by-product formation in yeast cell factories, and we experimentally validate two genes converting geraniol to geranial during geraniol biosynthesis. Cellular underground metabolism plays crucial roles in enzyme promiscuity, metabolism and biological evolution, but it has hardly been investigated. Here the authors combine retrobiosynthesis with deep learning enzyme annotation and enzyme–substrate prediction methods to explore it, reconstructing the yeast metabolic twin model, Yeast-MetaTwin.
|
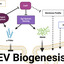






















For successful sorting, DE droplets must be significantly smaller (10–50 μm in diameter) than commercial FACS nozzles (typically 70–130 μm in diameter) while simultaneously large enough to encapsulate variants of interest (0.005–3 pL for bacteria to large mammalian cells, respectively) within the inner core volume (2–50 pL).