Your new post is loading...

|
Scooped by
?
December 22, 11:58 PM
|
Type I toxin-antitoxin (TA) systems are widely distributed in the prokaryotic world, known for their antitoxin RNA interfering with the cognate toxin mRNA translation. Beyond their canonical role in plasmid maintenance, these systems represent evolutionarily optimized regulatory modules with rapid kinetics and modular architectures. Here, we repurposed them as post-transcriptional regulatory RNA devices via reverse engineering. After isolating the core of type I TA pairs and demonstrating the independence between their structure and repression function, we reconstructed artificial TA pairs termed SRTS-OPRTS. A platform for generating orthogonal SRTS-OPRTS pairs with cross-species application (Bacillus subtilis, Escherichia coli, and Corynebacterium glutamicum) was developed by introducing structure and energy constraints. As an individual expressing element or a co-expressing 3′ UTR tag within specific mRNA, SRTS achieved quantitative regulation of the gene with 3′ UTR cognate OPRTS. Such portability enabled convenient construction of dynamic mutually inhibitory switches, where genes tagged by SRTS and OPRTS could regulate each other. Leveraging this approach, a selective lethal system was further constructed to enrich high-fluorescent mutants, resulting in up to 11.32-fold enhancement in mean fluorescence intensity. Overall, these synthetic RNA devices provide portable tools for gene regulation and offer a robust foundation for constructing dynamic genetic circuits.
|
|
Scooped by
?
December 22, 11:43 PM
|
Antimicrobial resistance (AMR) is one of the most pressing global health challenges, often referred to as a ‘silent pandemic.’ Towards this, ARKbase is developed as an integrated, curated, value-added knowledgebase for AMR, with focus on WHO Bacterial Priority Pathogens. ARKbase has three core modules namely—Database, Insight, and Comparative Analysis Module—each offering curated data, including data analysed with custom-built pipelines. The Database Module has curated reference genomes and pan genomes along with antimicrobial susceptibility testing (AST) profiles. The Insight Module has 14 sub-modules and offers deep annotations encompassing antimicrobial resistance genes, virulence factors, structure annotation, host–pathogen interactions, biosynthetic gene clusters, known antibiotics, protein–protein interactions, drug targets, co-targets, drug-target interactions, machine learning models, and curated transcriptomic datasets for antibiotic exposure. The Comparative Analysis module offers a simple interface for comparing antimicrobial resistance genes, virulence factors, and drug targets among different priority pathogens. The data included in ARKbase are quality-checked and curated as per CLSI standards for AST, EUCAST guidelines for genome sequence, and the FAIR data principles. ARKbase is the first comprehensive knowledgebase focusing on WHO priority pathogens and AWaRe classification, enabling a combined evidence approach towards a holistic understanding of AMR. Available at https://datascience.imtech.res.in/anshu/arkbase.
|
|
Scooped by
?
December 22, 10:53 PM
|
Nanopore sequencing enables direct, single-molecule interrogation of biopolymers and shows promise for analyzing not only DNA and RNA but also chemically modified bases, proteins, and other polymers. Expanded DNA alphabets, such as those found in xenonucleic acids (XNAs), open new possibilities for diagnostics, therapeutics, data storage, and engineered biology. However, robust sequencing strategies for these modified molecules remain lacking. While nanopore-based tools exist for some noncanonical bases, they often require extensive experimental calibration by measuring each base across many sequence contexts, which limits scalability and increases cost. In this work, we investigate computational methods for predicting the ionic current signals produced during nanopore sequencing of DNA containing noncanonical XNA bases, aiming to reduce the need for experimental calibration. We compare a sequence-based predictive model with two structure-aware approaches: one using graph-based molecular representations and another adapting a generative language model to molecular SMILES. Our findings show that while sequence context captures much of the signal variability, incorporating structural and chemical information improves predictive accuracy in specific cases. These results highlight the value of structural data representations and model design in scaling XNA sequencing, and suggest this framework could extend to modeling ionic currents from other complex biomolecules, such as proteins.
|
|
Scooped by
?
December 22, 4:18 PM
|
De novo protein design is advancing rapidly, but many targets remain inaccessible to current AI-based tools. Here we describe de novo designed globular domains that drive biomolecular condensation. Starting from a water-soluble, monomeric protein, we make variants with the same amino-acid composition but different surface-charge distributions: one with large patches of surface charge, and another with a more-homogeneous charge distribution. The individual domains form stable and discrete structures in solution, with the large-patch variant exhibiting more-attractive interprotein interactions. Next, two copies of each variant are joined with disordered linkers to generate dumbbell-like proteins. When expressed in eukaryotic cells, the large-patch variant forms intracellular puncta, whereas that with small patches does not. The assemblies are dynamic, liquid condensates in vitro and in cells. The structured domains facilitate functionalisation: we introduce fluorophore-binding sites to visualize fluorescent condensates directly in cells without a GFP reporter; and we manipulate the condensates using motor proteins.
|
|
Scooped by
?
December 22, 3:45 PM
|
Bacillus subtilis serves as a crucial host for industrial protein production, where the efficiency and regulation of its secretion system represent a central focus of applied research. Despite substantial genomic diversity among strains, current understanding of signal peptides, which are key elements in the secretion process, remains largely based on single model strains, lacking systematic investigation from a pan-genomic perspective. This study integrates pan-genomics and deep learning to construct a pan-secretome profile of 287 B. subtilis strains. Our analysis reveals an open repertoire of signal peptides in B. subtilis. Our results reveal an open pan-signal peptidome in B. subtilis. Core signal peptides exhibit high sequence conservation and primarily direct the localization of housekeeping proteins, whereas accessory signal peptides govern the extracellular secretion of environment-responsive proteins, forming a functional housekeeping-adaptive dichotomy. We demonstrated at a broad scale the widespread evolutionary decoupling between signal peptides and their corresponding mature peptides, uncovering a modular evolutionary mechanism in protein evolution. While the deep learning model achieved high accuracy (88.0%) in discriminating core and accessory gene-encoded full-length proteins, its performance was considerably limited when using signal peptide sequences alone (69.7% accuracy), reflecting the information constraints inherent to short sequences. This study provides a systematic portrayal of the signal peptide evolutionary landscape in B. subtilis at the pan-genome scale, advancing our understanding of the evolutionary principles governing bacterial secretion systems and establishing a theoretical foundation for optimizing industrial protein production through rational signal peptide design.
|
|
Scooped by
?
December 22, 3:21 PM
|
Predicting the assembly and stability of microbial ecosystems is fundamentally challenged by the stochasticity inherent in historical contingencies. Although these dynamics often appear reproducible at coarse taxonomic or functional scales, the mechanisms governing fine-grained species-level variation and the emergence of alternative stable states remain an unresolved problem in ecology. Bridging the gap between individual species variability and the emergence of predictable community-level attractors is essential for a mechanistic understanding of ecosystem resilience. Here, by analyzing replicate microbial communities assembled in vitro, we demonstrate that community structure is built upon individual species exhibiting robustly bimodal abundance distributions. Each taxon exists in either a high- or low-abundance state. We show that the collective configuration of these binary species states - a phenomenon also observed in natural systems - drives the correlation structure between community members and defines a set of discrete, alternative community-level attractors. By examining the prevalence of species across these attractors, we uncover a striking taxonomic signal: phylogenetically close relatives are significantly more likely to display reciprocal prevalence patterns - where the dominance of one relative necessitates the suppression of the other - than expected by chance. This discovery directly rejects models based on simple functional redundancy. Instead, it proves that state-dependent competitive exclusion is a primary driver of community divergence, where the identity of the ``winner'' is contingent on the collective state of the entire community. Our work reframes microbial community structure through the lens of discrete population states, providing a predictive framework for understanding ecosystem stability and the engineering of community functions.
|
|
Scooped by
?
December 22, 1:02 PM
|
Microplastics have emerged as major environmental hazards that require efficient, cost-effective, and sustainable remediation technologies. This study introduces an integrative platform for the remediation and upcycling of microplastics by algae, while synergizing with plastic upcycling, wastewater treatment, and algal production. The strategy employs a mechanism that enhances hydrophobic interactions between the cell surface and microplastics, enabling rapid aggregation and removal. The platform achieves a superior microplastic removal efficiency of 91.4% within 1 hour, with a capacity of 0.1-gram microplastic per gram of biomass. Furthermore, the study demonstrates an upcycling strategy that converts microplastics-enriched cyanobacteria into plastic composites with unique performance. This work also integrates microplastic removal with cyanobacterial bioproduction and wastewater treatment, offering an approach that synergizes remediation with these value-added processes. Ultimately, this platform provides a viable and sustainable pathway to address microplastic pollution by creating value through plastic upcycling, wastewater nutrient removal, and CO2-based bioproduction. Microplastics (MPs) represent an environmental hazard which must be resolved by efficient, cheap, and sustainable remediation technology. Here the authors use an engineered algae to capture MPs and treat wastewater, the captured algae-plastic mix is upcycled into a tougher bioplastic composite.
|
|
Scooped by
?
December 22, 11:45 AM
|
Microplastic biofilms, known as the “plastisphere”, harbor diverse microbial communities and serve as reservoirs for antibiotic resistance genes (ARGs). This review discussed the mechanisms driving bacterial community alteration on microplastics and delineated the pathways through which ARGs transfer within microplastic biofilms. We expected to provide a comprehensive understanding of the ecological and human health impacts associated with microplastic biofilms and ARGs, thereby informing strategies to mitigate plastic pollution and its risks.
|
|
Scooped by
?
December 21, 11:07 PM
|
Plants rely heavily on a complex innate immune system to repel microbial attacks, and antimicrobial peptides (AMPs) play a crucial role as central immune modulators of their immune response. These small, structurally diverse molecules exhibit broad-spectrum activities against bacteria, fungi, viruses, and nematodes through direct membrane disruption, interference with intracellular targets, and modulation of host signaling networks. Recent progress in multi-omics technologies, including genomics, transcriptomics, proteomics, and metabolomics, combined with synthetic biology, has led to a significant increase in our understanding of classification, biosynthetic pathways, structure-function relationships, and the regulatory integration of AMPs within pattern-triggered immunity (PTI) and effector-triggered immunity (ETI). Some of these latter tools accelerate rational peptide design and enable applications in agriculture: among others, transgenic crops possessing constitutive or pathogen-inducible AMP expression, as well as peptide-based agrochemicals, represent eco-friendly alternatives to classical pesticides. Despite such breakthroughs, major challenges persist that currently limit large-scale deployment: rapid pathogen evolution of resistance, insufficient target specificity, peptide instability under field conditions, potential phytotoxicity, and complex regulatory approval pathways. Such barriers will require integrated systems-biology approaches, improved delivery platforms, for example, nanotechnology or bioencapsulation, and precise engineering of AMP-host interactions. This review consolidates current information on plant AMPs, highlighting transformative multi-omics insights and critically assessing the remaining hurdles to be overcome for the full utilization of AMPs in next-generation sustainable crop protection and global food security.
|
|
Scooped by
?
December 21, 1:07 PM
|
Antibody-based therapies have transformed the management of immune-mediated inflammatory diseases (IMIDs), but the need for frequent injections often leads to inadequate patient adherence and suboptimal long-term disease control. To address this challenge, we develop AIDEN (aid for IMIDs: engineered EcN), an engineered probiotic platform that enables oral delivery of therapeutic antibodies using synthetic biology. In this study, we assess the efficacy of AIDEN-IL17, a variant designed to secrete single-chain variable fragments targeting interleukin-17A (IL-17A), in murine models of psoriasis and inflammatory bowel disease. AIDEN-IL17 exhibits stable gut colonization and sustained in situ antibody production, resulting in moderate reduction of systemic IL-17A levels and significant amelioration of disease symptoms. Notably, the AIDEN platform is modular and adaptable for delivering a broad range of antibody therapeutics, offering a promising, patient-friendly strategy for the treatment of IMIDs.
|
|
Scooped by
?
December 21, 12:54 PM
|
Cell-to-cell variability often limits the efficiency of microbial bioproduction, yet how individual cells fluctuate over time and how these fluctuations shape population-level output remain unclear. To address this issue, we tracked a heterologous betaxanthin pathway in Escherichia coli using microfluidics-assisted time-lapse microscopy, allowing simultaneous measurement of fluctuations in betaxanthin, its biosynthetic enzyme DOD and growth across generations. Here we show that over 50% of high betaxanthin producers become medium or low producers after two divisions. Betaxanthin variation primarily originates from DOD noise, with a smaller contribution from growth rate fluctuations. We further develop a stochastic model to explore various control circuits and find that pathway enzyme or metabolite-based growth selection strategies are most effective in enhancing production. We experimentally validate the model by coupling enzyme expression to nutrient availability, which enriches high producers and boosts titer by 4.4-fold. Our results highlight key sources of metabolic heterogeneity and provide a framework for designing robust microbial processes. Cell-to-cell variability limits efficient microbial production. Here, the authors track single cells to reveal enzyme noise as the main source of bioproduction variation, and by coupling growth to pathway performance, they selectively enrich high producers and substantially boost overall titres.
|
|
Scooped by
?
December 21, 11:19 AM
|
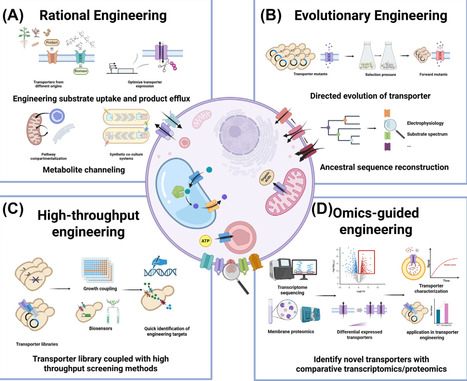
Membrane transporters play crucial roles in metabolite exchange, cellular communication, and metabolic homeostasis and are attractive targets in metabolic engineering for the development of microbial cell factories. While transporter engineering has proven effective in enhancing nutrient uptake, improving product secretion, and optimizing metabolic flux, its broader application is limited by incomplete knowledge of membrane transport systems and the sometimes high promiscuity of transporters, often resulting in unpredictable outcomes. This review provides an overview of recent progress in transporter engineering and characterization methods, highlighting their potential to enhance the production of bio-based chemicals.
|
|
Scooped by
?
December 20, 11:46 PM
|
Synthetic microbial communities (SynComs) are microbial consortia with defined taxonomic and functional traits, so that the combination elicits a predictable response under defined conditions. SynComs are artificially designed to enable inter-species metabolic interactions, metabolic division of labor, and ecological interactions that can elicit phenotypes like colonization stability and environmental adaptation. As an applied tool, SynComs have been deployed in diverse contexts, including agriculture, industry, and environmental ecology. This systematic review explores the processes used to construct SynComs, the mechanisms of metabolic interaction between members, and a review of the different ways that SynComs have been applied. We also explore the challenges for SynCom development and application, and future research directions that could overcome these challenges. SynComs are a powerful tool in our arsenal of applied technologies, but research and application are still nascent. While advances have been made, more research is needed to ensure SynCom technologies do not threaten global ecological security. SynCom technology represents a versatile platform for the controlled manipulation of microbial systems, enabling targeted modification of ecological and physiological processes. This emerging field marks a transition from descriptive biology toward a predictive and engineering-driven framework for understanding and shaping living systems.
|
|
|
Scooped by
?
December 22, 11:46 PM
|
Protein structure prediction has a long history of benchmarking efforts such as critical assessment of structure prediction, continuous automated model evaluation and critical assessment of prediction of interactions. With the rise of artificial intelligence-based methods for prediction of macromolecular complexes, benchmarking with large datasets and robust, unsupervised scores to compare predictions against a reference has become essential. Also, the increasing size and complexity of experimentally determined reference structures by crystallography or cryogenic electron microscopy poses challenges for structure comparison methods. Here we review the current state of the art in scoring methodologies, identify existing limitations and present more suitable approaches for scoring of tertiary and quaternary structures, protein–protein interfaces and protein–ligand complexes. Our methods are designed to scale efficiently, enabling the assessment of large, complex systems. All developments are available in the structure benchmarking framework of OpenStructure. OpenStructure is open source software and available for free at https://openstructure.org/ . This study advances methods for benchmarking macromolecular complex predictions by introducing a scalable open-source framework used in recent community assessments to compare structures, interfaces and ligand interactions against experimental data.
|
|
Scooped by
?
December 22, 10:58 PM
|
CRISPR-cas provide sequence-specific mechanisms for targeting foreign DNA or RNA and have been widely used in genome editing and DNA detection. Type V CRISPR–Cas systems are characterized by a single RNA-guided RuvC domain-containing effector, Cas12. Here, through comprehensive mining of large-scale genomic and metagenomic data from microbial sources, we identified a new Class 2 CRISPR–Cas effector superfamily, designated Casδ, comprising three members with protein sizes ranging from 867 to 936 amino acids. Biochemical analyses revealed that Casδ-1 functions as a single RNA-guided endonuclease with specific recognition of 5′-RYR-3′ protospacer-adjacent motifs, where R represents A or G, and Y represents T or C. Casδ-1 exhibits robust double-stranded DNA cleavage activity and target-dependent trans-cleavage activity. Casδ-1 mediates efficient genome editing across species, achieving up to 60% indel rates in human cells while generating homozygous knockout lines in two agriculturally important monocot species (Oryza sativa and Zea mays) through stable transformation. Structural and evolutionary analyses reveal Casδ as an evolutionary transitional nuclease bridging Cas12n and canonical type V systems, featuring a C-terminal loop that is essential for activity. Collectively, Casδ is an evolutionarily distinct, compact (<1000 aa), tracrRNA-free CRISPR system enabling versatile cross-kingdom genome editing.
|
|
Scooped by
?
December 22, 9:55 PM
|
Transfer RNA (tRNA) modifications tune translation rates and codon optimality, thereby optimizing co-translational protein folding. However, the mechanisms by which tRNA modifications modulate codon optimality and trigger phenotypes remain unclear. Here, we show that ribosomes stall at specific modification-dependent codon pairs in wobble uridine modification (U34) mutants. This triggers ribosome collisions and a coordinated hierarchical response of cellular quality control pathways. High-resolution ribosome profiling reveals an unexpected functional diversity of U34 modifications during decoding. For instance, 5-carbamoylmethyluridine (ncm5U) exhibits distinct effects at the A and P sites. Importantly, ribosomes only slow down at a fraction of codons decoded by hypomodified tRNA, and the decoding speed of most codons remains unaffected. However, the translation speed of a codon largely depends on the identity of A- and P-site codons. Stalling at modification-dependent codon pairs induces ribosome collisions, triggering ribosome-associated quality control (RQC) and preventing protein aggregation by degrading aberrant nascent peptides and messenger RNAs. Inactivation of RQC stimulates the expression of molecular chaperones that remove protein aggregates. Our results demonstrate that loss of tRNA modifications primarily disrupts translation rates of suboptimal codon pairs, showing the coordinated regulation and adaptability of cellular surveillance systems. These systems ensure efficient and accurate protein synthesis and maintain protein homeostasis.
|
|
Scooped by
?
December 22, 4:06 PM
|
In nature, complex multicellular structures originate from individual cells containing all essential information for differentiation, patterning and morphogenesis. Synthetic biology enables a bottom-up approach to study these processes by engineering and combining individual modules to progressively increase the system's complexity. Here, we engineered a multi-step differentiation program in the model prokaryote E. coli. Starting from genetically identical cells and without providing any external positional information, we generated autonomous spatial patterns of colonies on a solid surface. We first employed a toggle switch to break population homogeneity (symmetry breaking), stochastically differentiating cells into two subpopulations: senders and receivers. Next, we enabled further differentiation of receiver colonies located in close proximity to sender colonies via quorum-sensing based communication (paracrine signaling). Finally, the newly emerged population matured into a different cell type via an orthogonal, self-activating, quorum sensing signal (autocrine signaling). The diversity of spatial patterns generated by this multi-step program was accurately captured by simulations of a corresponding mathematical model. Together, these results demonstrate that multi-step differentiation programs can be engineered in unicellular bacteria to drive fully self-organized spatial pattern formation.
|
|
Scooped by
?
December 22, 3:40 PM
|
Microorganisms employ diverse metabolic strategies to cope with environmental variability, yet the evolutionary conditions that favor specialists, hierarchical utilizers, or co-utilizers remain poorly understood. Here, we investigate how fluctuations in resource supply ratios drive the evolution of metabolic strategies. By simulating microbial community assembly in environments with variable resource ratios, we show that diverse metabolic strategies emerge as evolutionary optima in different fluctuation regimes. We find that environments with balanced resource supply select for specialists, whereas imbalanced environments select for generalists. As the magnitude of fluctuations in resource supply ratios progressively increases, generalist strategies smoothly shift from those of hierarchical utilizers to co-utilizers. We identify the duration of temporal niches as the mechanistic driver that selects for reduced lag times over maximal growth rates. These findings demonstrate how the temporal structure of resource availability can spontaneously generate the three distinct metabolic strategies observed in nature.
|
|
Scooped by
?
December 22, 2:14 PM
|
Microplastics are widely recognized as persistent and pervasive contaminants that endanger human health and ecosystems. Traditional remedial techniques are problematic due to high costs and inefficiency. One sustainable method of dissolving tough polymers into recyclable parts is through microbial and enzymatic engineering. Recent advances in genome-editing technologies, enzyme redesign, and synthetic biology particularly CRISPR-based systems have transformed the way we approach enhancing the efficiency of biodegradation. Recent CRISPR applications, such as base editing and promoter modification, have improved the stability and expression of enzymes, accelerating the catalytic activity of PET hydrolases, including PETase and cutinase. To enable scalable plastic biodegradation, this review combines hybrid CRISPR-based systems with microbial and enzyme engineering techniques. The goals of computational and machine learning–based enzyme design is thermostability and substrate adaptation, while hybrid microbial communities made up of modified bacteria and fungi improve degradation through cooperative processes. Furthermore, combining synthetic biology with hybrid remediation techniques, such as biofilm reactors and enzyme-nanoparticle conjugates, links laboratory research developments with real-world applications. However, issues remain regarding the scalability of polyethylene (PE) and polystyrene (PS) degradation, biosafety standards for genetically modified organisms (GMOs), and environmental hazards associated with degradation byproducts. To effectively manage plastic waste, future research should focus on creating thermostable enzymes, forming synthetic consortia guided by multi-omics, and developing safe hybrid bio-physical systems that support circular bio economy models.
|
|
Scooped by
?
December 22, 12:19 PM
|
Biosensors based on transcription factors (TFs) have shown extensive applications in synthetic biology. Due to the complex multi-domain structure of effector-TF-DNA, computational design of TFs remains a challenge. Here, we present the successful structure-guided computational design of the access tunnel, ligand binding, allosteric transition process for an allulose-responsive PsiR. It enables a 20-fold increase in sensitivity, reducing the EC50 of PsiR-allulose biosensors (PABs) from 16 mM to 0.8 mM, and delivers a PAB box possessing the detection range from 10 μM to 100 mM. We further validate its broader applicability in enhancing sensitivity of LacI-IPTG biosensor. Based on the developed PABs, we present the inducer-free allulose-mediated auto-inducible protein expression system, and demonstrate an allulose-triggered CRISPR interference circuit for dynamic metabolic regulation. It facilitates a 68% increase in allulose titer and achieves a high yield of 0.43 g/g glucose. This work provides the versatile TF toolbox for developing allulose-triggered regulation circuits in biotechnology application. Transcription-factor biosensors enable programmable metabolism. Here, the authors integrate structure and computation for mechanism-guided iterative redesign of the allulose biosensor PsiR, increasing sensitivity and dynamic range, enabling allulose-triggered expression system and CRISPRi circuit.
|
|
Scooped by
?
December 22, 11:10 AM
|
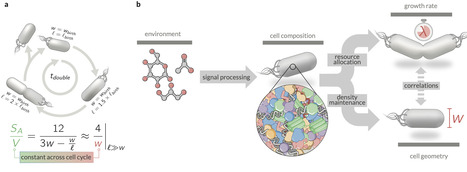
Microbes precisely control their composition and geometry across diverse growth conditions, yet the mechanisms coordinating these processes remain unclear. Here, we integrate quantitative proteomics, microscopy, and biochemical measurements to reveal a biophysical principle linking these properties in Escherichia coli: cytoplasmic and membrane protein densities maintain a tightly conserved ratio across growth conditions, while the periplasmic density varies. Building on this observation, we develop a mathematical model demonstrating that maintaining this density ratio constrains the surface-to-volume ratio as a nonlinear function of proteome composition, specifically the ribosomal proteome fraction and partitioning between cellular compartments. The model holds under guanosine tetraphosphate perturbations that alter ribosome levels, further demonstrating that cellular geometry is not strictly determined by growth rate. These findings provide a biophysical framework for geometry control, underscoring density maintenance as a key physiological constraint that shapes cellular phenotypes. Chure et al. analyse experimental data to show that E. coli bacteria maintain stable protein density ratios between cytoplasm and membranes. In addition, they develop a biophysical model that predicts surface-to-volume ratio from ribosomal content and protein partitioning across cell compartments.
|
|
Scooped by
?
December 21, 1:28 PM
|
Over the next decade, mammalian synthetic biology should become the first precision control layer for human physiology. Synthetic gene circuits will sense molecules—glucose, lipids, and cytokines—and actuate closed-loop, real-time corrections integrated with host metabolism. This retires open-loop, Paracelsus-era dosing—pills at fixed intervals—in favor of continuous control. Placement should follow safety and access, not organ identity: subcutaneous sensing and control, by autologous cells, exosome/RNA cargoes, or xenogeneic tissue, and encapsulation or immune shielding manage risk. Metabolic control is modular—blood glucose does not need be controlled from the pancreas, nor do lipids need to be lowered by the liver. As circuits interface with metabolism, diagnosis, prevention, and therapy converge—shifting care from chronic management to cure.
|
|
Scooped by
?
December 21, 1:01 PM
|
Vaccines are the most effective tool in preventing and managing infectious diseases. One of the critical challenges in vaccine development is the selection of suitable target antigens from the thousands of proteins produced by pathogens. Artificial intelligence is anticipated to play a significant role in addressing this challenge. In this study, we develop a framework termed PLGDL for protective antigen prediction that employs Protein Language and Geometric Deep Learning models. This framework leverages both primary sequence features and three-dimensional structural features of protein antigens, thereby reducing the biases associated with manually curated features. Our integrated model exhibits robustness across both constructed and public datasets and is applicable to viruses, bacteria, and eukaryotic pathogens. Notably, when applied to the ongoing Mpox outbreak, our model not only quickly identifies multiple known antigens but also discovers a protective antigen: G10R. Here, our study provides a high-performance screening tool for protective vaccine antigen prediction by synergistically utilizing the capabilities of protein language and geometric deep learning models, providing substantive insights and methodological advancements for rapid vaccine development. Vaccines are the most effective tool in managing infectious disease and characterizing features of protective epitopes could help in prediction methods. Here the authors use protein language and geometric deep learning frameworks to investigate primary sequence features and structural features to identify and predict potential antigens, showing prediction of a protective mpox epitope using this method.
|
|
Scooped by
?
December 21, 12:29 PM
|
Heterogeneity within clonal cell populations remains a critical bottleneck within bioprocess engineering, notably by undermining bioproduction yields. Efforts to mitigate its impact have, however, been hampered by technological difficulties quantifying metabolism at the single-cell level. Here, we propose a framework based on single-cell biosensor analysis that enables robust characterization of cell’s metabolic states, leveraging it to detect and isolate isogeneic heterogeneity in response to environmental perturbations and within microbial cell factories. We identify acute and gradual glucose depletion to induce differentiation of metabolically distinct subpopulations and reveal these subpopulations to exhibit differential production capabilities, with lower intracellular pH subpopulations exhibiting enhanced product accumulation within violacein-producing strains but reduced yields within lycopene-producing strains. Lastly, we highlight galactose cultivation as a method to modulate subpopulation dynamics towards higher-producing lycopene phenotypes. Altogether, our research provides insights into subpopulation differentiation and establishes promising avenues for the engineering of more robust and higher-producing strains. Heterogeneity within clonal cell populations affects bioprocess engineering. Here, the authors report a biosensor-based toolkit to investigate phenotypic heterogeneity in engineered yeast, reveal pH-based subpopulations and metabolite production states, and modulate/shift subpopulation dynamics to increase lycopene production.
|
|
Scooped by
?
December 20, 11:56 PM
|
Bacteria exhibit two lifestyles: planktonic free-floating individual cells or sessile multicellular aggregates known as biofilms. The biofilm lifecycle is characterized by three distinct stages: attachment, maturation and dispersal. Specific signals govern each stage triggering responses that spatially and temporally regulate bacterial attachment to a surface, synthesis of extracellular matrix components and their subsequent degradation. Characterizing these signals is therefore a valuable approach to develop novel antibiofilm therapies. Here, we used the model biofilm-forming bacterium Pseudomonas aeruginosa PAO1 to characterize the transcriptional profiles of each stage of the biofilm life cycle: attachment, biofilm maturation and spontaneous dispersal. We report that surface attachment was accompanied by the upregulation of genes comprising the mechanosensor Pil-Chp, whereas biofilm maturation characterised with the upregulation of genes involved in Pel polysaccharide synthesis, siaD and PA4396 diguanylate cyclases as well as pipA, fimX and PA5442. In contrast, dispersing cells upregulated genes responsible for alginate, rhamnolipid, and extracellular nucleases (eddA, eddB) biosynthesis, as well as the transcriptional regulator of dispersal amrZ. Additionally, genes encoding the quorum sensing dispersal molecule cis-2-decenoic acid (dspS and dspI), canonical phosphodiesterases (nbdA and rbdA) and eleven other c-di-GMP–related enzymes were also upregulated during dispersal. Our comprehensive analysis of transcriptional profiles associated with different biofilm stages allowed us to define a subset of fourteen genes as biomarkers of biofilm dispersal. Our study therefore provides benchmarking stage-specific transcriptional profiles for P. aeruginosa biofilms in closed culture systems, which led to the identification of a dispersal fingerprint marking the onset of dispersal.
|