 Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 7:46 PM
|
Bacterial RNA polymerase binds and unwinds promoter DNA to initiate transcription. The process is often inefficient and can be stimulated by activator proteins. For example, many activators bind RNA polymerase and promoters simultaneously, stabilizing their interaction. Working with the multiple antibiotic resistance activator (MarA) protein of E. coli, we have discovered an alternative mechanism. We show that, when bound upstream of the flgBCDEFGHIJ/KL operon, MarA perturbs base pairing adjacent to its DNA target. This compensates for inefficient duplex unwinding by RNA polymerase and, as a result, activates transcription. Consistent with our model, an appropriate base pair mismatch mimics the effect of, and removes the need for, MarA. As many regulators alter DNA conformation, we suggest that this mechanism of activation could be commonplace.
|
|
Scooped by
mhryu@live.com
Today, 7:36 PM
|
Precise modeling of transcriptional regulation is essential for the rational design of genetic circuits in synthetic biology. Current computational approaches for predicting transcriptional activity (ITX) typically lack mechanistic clarity, composability, and scalability, and require extensive training data. Here, we present a modular thermodynamic modeling framework that explicitly parameterizes molecular interactions among promoters, RNA polymerase (RNAP) and transcription factors (TFs). Implemented as the computational platform, T-Pro, this approach provides robust interpretability, scalability, and predictive power. Experimental validation across three distinct bacteria—Escherichia coli, Bacillus subtilis, and Corynebacterium glutamicum—demonstrates substantial improvements (up to 20-fold) in a composite transcriptional performance metric (Fmax*FC), achieved within only three Design–Build–Test–Learn cycles and fewer than five genetic constructs in total. Furthermore, we validate the framework by engineering multispecies bacterial communication circuit, highlighting its broad utility and generalizability. The principles and tools developed here thus enable efficient, rational optimization of transcriptional regulation across diverse prokaryotic hosts.
|
|
Scooped by
mhryu@live.com
Today, 7:18 PM
|
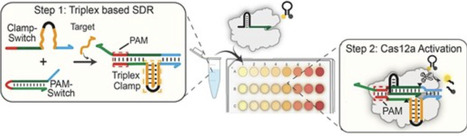
We present a molecular strategy that enables the programmable activation of the CRISPR–Cas12a system in response to triplex DNA formation triggered by single-stranded DNA (ssDNA) or RNA inputs. Our triplex-controlled Cas12a assay leverages the high specificity of clamp-like triplex structures to control a toehold-based strand displacement reaction within a rationally designed DNA hairpin (PAM-Switch). Upon displacement and protospacer adjacent motif (PAM) complementation, the Cas12a ribonucleoprotein (RNP) is activated, initiating trans-cleavage and producing a concentration-dependent fluorescent signal. By decoupling target recognition (via triplex formation) from direct hybridization with the Cas12a–crRNA complex, the assay eliminates the need for target-specific crRNAs. This design also allows multiple detection of distinct nucleic acid (NA) targets using a single Cas12a reaction mix. Through the use of triplex-based clamps, the proposed platform achieves enhanced specificity for single-nucleotide variants and supports the detection of both ssDNA and RNA targets across a broad range of lengths (10–20 nucleotides), addressing key limitations in current Cas12a-based diagnostics and opening new avenues for NA sensing.
|
|
Scooped by
mhryu@live.com
Today, 7:11 PM
|
The Encyclopedia of Domains (TED) provides domain annotations for proteins in the AlphaFold Protein Structure Database (AFDB) using a consensus of three state-of-the-art structure-based methods. We used these TED domain annotations to construct profile Hidden Markov models (HMMs), collectively forming the TED Library of HMM (TEDLH). TEDLH enables sensitive sequence and profile searches, supporting systematic exploration of protein domain families and their evolutionary relationships. TEDLH links domain HMMs to experimentally determined CATH-PDB structures through direct (primary) and transitive (secondary and tertiary) relationships. Fewer than half of TEDLH HMMs are directly linked to a CATH-PDB domain; the remaining models are connected through transitive relationships. These transitive links extend coverage into more divergent regions of sequence space and better represent CATH superfamily diversity. HMM-HMM comparisons within CATH superfamily 3.30.70.100 illustrate how transitive relationships expand sequence coverage in TEDLH. In this superfamily, 4,813 TEDLH HMMs are connected to 212 CATH-PDB representatives. Primary, secondary, and tertiary relationships progressively capture more divergent sequences (pairwise sequence identity <20%) that retain structural similarity (TM-score >0.6) and a conserved two-layer α/β sandwich core fold. All-against-all HMM-HMM comparisons across TEDLH also reveal sequence similarities across the CATH hierarchy (cross-hits). At low query coverage (<50%), cross-hits are more frequent between CATH classes, whereas at higher coverage thresholds (>70%) they predominantly occur between superfamilies. These cross-hits are not driven by superfamily size or sequence diversity and can provide guidance for CATH curation. As an example, analysis of cross-hits between superfamilies 2.170.130.30 and 3.10.20.30 reveals evolutionary relationships between these groups.
|
|
Scooped by
mhryu@live.com
Today, 5:35 PM
|
Antifungal resistance is a growing crisis affecting human health and agriculture that could be accelerated by global change. We call for a One Health research agenda to systematically investigate environmental drivers and inform policy to mitigate the potential of global environmental change in fuelling antifungal resistance.
|
|
Scooped by
mhryu@live.com
Today, 4:57 PM
|
Metabolites generated by host and pathogen have a major impact on the severity and outcomes of infection. The metabolic response to infection shapes the nature and intensity of the immune response, both in bloodstream infections and, especially, in the pathogenesis of pneumonia. Some metabolites are closely linked to pro-inflammatory responses, whereas others act as immunomodulators in mitigating damage to the host, a common consequence of inflammation. Immunometabolites are also major factors in driving bacterial adaptation to the host, enabling pathogens acquired from environmental sources to modify their gene expression to optimize for persistent infection. In this era of diminishing antimicrobial efficacy, an appreciation of the immunometabolic responses to bacterial infection may provide novel targets for therapy.
|
|
Scooped by
mhryu@live.com
Today, 4:32 PM
|
Auto-brewery syndrome (ABS) is a rarely diagnosed disorder of alcohol intoxication due to gut microbial ethanol production. Despite case reports and a small cohort study, the microbiological profiles of patients remain poorly understood. Here we conducted an observational study of 22 patients with ABS and 21 unaffected household partners. Faecal samples from individuals with ABS during a flare produced more ethanol in vitro, which could be reduced by antibiotic treatment. Gut microbiome analysis using metagenomics revealed an enrichment of Proteobacteria, including Escherichia coli and Klebsiella pneumoniae. Genes in metabolic pathways associated with ethanol production were enriched, including the mixed-acid fermentation pathway, heterolactic fermentation pathway and ethanolamine utilization pathway. Faecal metabolomics revealed increased acetate levels associated with ABS, which correlated with blood alcohol concentrations. Finally, one patient was treated with faecal microbiota transplantation, with positive correlations between gut microbiota composition and function, and symptoms. These findings can inform future clinical interventions for ABS. Gut microorganisms capable of producing ethanol cause alcohol intoxication, which can be corrected via faecal microbiota transplantation.
|
|
Scooped by
mhryu@live.com
January 7, 11:54 PM
|
RNA secondary structures serve as a bridge between RNA sequences and often-unknown three-dimensional structures, offering insights into base pairing, structural motifs, and the overall organization of RNA molecules. To support efficient visualization and editing of these structures, we present Exornata, a modern, web-based editor designed to facilitate generation of detailed and standardized RNA secondary structure modeling. Exornata, is an expanded successor to the original XRNA software and is implemented using React and JavaScript/TypeScript technologies to ensure flexibility, interactivity, and high-quality rendering. Users can load, edit, and export RNA structures in multiple supported formats, ranging from conventional SVG to an advanced, in-house–developed JSON schema designed for interoperability with other resources such as R2DT, Traveler, and RiboVision2. The application supports multiple constraint-based editing modes (e.g. nucleotide, strand, helix, and domain) allowing precise and hierarchical manipulation of RNA elements. Exornata supports detailed, interactive visualization of canonical and non-canonical base pairs; it enables researchers to format and annotate structures, and to integrate them into broader bioinformatics pipelines. The tool is open-source, freely available at https://ldwlab.github.io/XRNA-React/, and is accompanied by a user guide.
|
|
Scooped by
mhryu@live.com
January 7, 11:11 PM
|
The ability to generate genomic diversity expands opportunities for understanding and engineering biology. Here, we demonstrate on-demand generation of diversity in bacterial genome configurations and its application to probing physiology under altered genome organization. We engineered the fast-growing bacterium Vibrio natriegens to enable large-scale stochastic duplications, translocations, inversions, and deletions, producing populations with a wide range of genome arrangements. We investigated phenotypic robustness to genome reconfigurations and found that distinct genome organizations can support stable physiology, indicating that bacteria may tolerate chromosomal alterations more readily than previously appreciated. Our work provides a framework for advancing the understanding and engineering of bacterial genomes and suggests that genome reconfigurations may contribute to phenogenetic drift, allowing for evolutionary exploration while preserving phenotype.
|
|
Scooped by
mhryu@live.com
January 7, 5:08 PM
|
Engineering DNA polymerases to efficiently synthesize artificial or noncognate nucleic acids remains an essential challenge in synthetic biology. Here we describe an evolutionary campaign designed to convert a family of highly selective DNA polymerases into an unnatural homolog with strong RNA synthesis activity. Starting from a homologous recombination library, a short evolutionary path was achieved using a single-cell droplet-based microfluidic selection strategy to produce C28, a newly engineered polymerase that can synthesize RNA with a rate of ~3 nt s−1 and of >99% fidelity. C28 is capable of long-range RNA synthesis, reverse transcription and chimeric DNA–RNA amplification using the PCR. Despite strong discrimination against other genetic systems, C28 readily accepts several 2′F and base-modified RNA analogs. Together, these findings highlight the power of directed evolution as an approach for reprogramming DNA polymerases with activities that could help drive future applications in biotechnology and medicine. Engineering polymerases to synthesize alternative genetic polymers remains a challenging problem in synthetic biology. Using DNA shuffling and droplet microfluidics, the current study provides a short evolutionary path from a DNA polymerase to one with robust RNA-synthesizing activity.
|
|
Scooped by
mhryu@live.com
January 7, 4:32 PM
|
Viable but non-culturable (VBNC) cells represent a reversible, metabolically active state that promotes the survival of bacteria under stressful conditions and their persistence in healthcare facilities and food industry. We conducted a systematic review following PRISMA 2020 guidelines to identify in vitro methodologies for inducing and resuscitating VBNC Enterobacteriaceae and Pseudomonas aeruginosa, and to determine key influencing factors. Eligible studies reported in vitro resuscitation of these species. Searches were performed in MEDLINE (PubMed), Scopus, and Google Scholar up to July 2025. Two independent reviewers screened and extracted data. Exclusion criteria included absence of original experimental data, focus on other species, or lack of clear VBNC definition. Risk of bias was qualitatively assessed. Analyses were descriptive without meta-analysis. Of the 1041 records, 24 articles (27 studies) were included. Resuscitation protocols typically employed standard culture media with additives and moderate incubation temperatures, with most successful recoveries occurring after 24–48 h. P. aeruginosa generally required less supplementation than Enterobacteriaceae. Reported mechanisms involved metabolic reactivation, oxidative stress modulation, nutrient sensing, and ribosome reactivation. The limitations of our study include protocol heterogeneity, lack of standardization, and selective reporting. While simple resuscitation methods were often effective, tailoring conditions to species-specific ecological preferences appears critical. Standardized approaches of VBNC cells will improve detection, risk assessment, and infection control.
|
|
Scooped by
mhryu@live.com
January 7, 4:24 PM
|
Site-specific DNA recombinases are powerful tools for genome engineering. The recent convergence of DNA recombinase-mediated technology with prime editing has established a promising new frontier for large-scale genomic integration. However, this strategy has limitations, including low intrinsic catalytic efficiency of DNA recombination enzymes and inefficiency of their long DNA landing pad insertions. To overcome these challenges, we developed a novel directed evolution strategy using progressively shortening landing pads as a selective pressure. The resulting evolved variants, VK and AVK, exhibited substantially enhanced intrinsic activity that is maintained on short-length landing pads. This enables a powerful synergistic effect with increased insertion efficiency of prime editing, leading to a dramatic increase in overall integration efficiency while maintaining high genomic specificity. This work thus provides a new class of highly efficient recombinases and a robust engineering strategy with broad applicability for both the development of gene therapies and fundamental research.
|
|
Scooped by
mhryu@live.com
January 7, 4:14 PM
|
As the number and complexity of RNA and DNA structures continue to expand, there is a growing need for robust yet accessible tools that support their accurate interpretation, validation, and refinement. We present DNATCO v5.0 https://dnatco.datmos.org/app an interactive web application for comprehensive structural analysis of nucleic acids. DNATCO integrates the NtC dinucleotide conformational classes and the CANA structural alphabet to provide an intuitive, geometrically complete description of local backbone and base orientations, complemented by interactive visualization of base pairing. The platform performs quantitative validation of conformational similarity and covalent bond lengths and angles, using newly established nucleic-acid valence-geometry standards. Quantitative validation encompasses the confal score and scattergrams mapping the fit between experimental electron density and geometry similarity to the closest NtC class. All outputs are downloadable. Integrated diagnostic tools help users identify unusual or problematic regions, explore alternative conformations, and generate torsion-restraint files for downstream. DNATCO v5.0 is implemented entirely client-side via WebAssembly, ensuring fast performance and preserving data privacy, and supports both PDB and user-provided structural models. By combining a rigorous geometric framework with an approachable interface, DNATCO enables both non-experts and specialists to evaluate nucleic-acid structures with greater confidence and to improve models in ways that support accurate biological interpretation.
|
|
|
Scooped by
mhryu@live.com
Today, 7:40 PM
|
The messenger RNA 3' untranslated region contains important regulatory sequences, including upstream sequence elements (USEs), which regulate gene expression. One well-characterised USE in the 3'UTR of the Drosophila polo gene affects adult fly phenotypes when disrupted. We have now identified a highly conserved sequence within this USE (DplUSE) in the 3'UTR of several vertebrate genes, including in zebrafish, mouse, and human genomes and show that DplUSE enhances gene expression in human cells and zebrafish embryos. We show that, in humans, DplUSE-containing genes are associated with congenital disease processes, and that disruption of DplUSE function impairs zebrafish development. We also found that HuR/ELAVL1, hnRNPC, and PTBP1/hnRNPI bind to DplUSE RNA and are required for its activity in a human cell line, suggesting a highly conserved mechanism across distantly related species. Our results indicate that PTBP1 has a global function in alternative polyadenylation, activating the selection of distal polyA sites and repressing intronic polyadenylation in DplUSE-containing genes while hnRNPC and HuR modulate their expression. Additionally, we found that a colon cancer-associated SNP in the POU2AF2/C11orf53 3'UTR creates an ectopic DplUSE site, increasing gene expression in zebrafish gut cells and in a human cell line. We have therefore identified a short 3'UTR motif present in diverse vertebrate genes that controls their expression through conserved RBPs interactions and is implicated in human disease.
|
|
Scooped by
mhryu@live.com
Today, 7:24 PM
|
The expansion of the genetic alphabet through the development of unnatural base pairs (UBPs) has the potential to revolutionize synthetic biology and biotechnology. However, the replication of the UBPs by eukaryotic DNA polymerases is largely unexplored and the sequencing of the UBPs remains challenging. Herein, we explored and demonstrated the activity of human DNA polymerase β (Pol β) for the efficient and specific synthesis and extension of a panel of representative UBPs, including dNaM–dTPT3, dCNMO–dTPT3, and their functionalized derivatives. Based on this, we established a method for the sequencing of DNAs containing different unnatural bases, involving stalled primer extension mediated by Pol β, selective conversion of an unnatural nucleotide into two different natural ones in parallel and further primer extension mediated by Taq DNA polymerase, and Sanger or deep sequencing of the produced natural DNAs to locate the unnatural bases. The precision, universality, and potential for high-throughput applications of this method were demonstrated by the successful sequencing of various DNAs containing one or multiple of different unnatural bases. This work suggests the possibility of integrating the UBPs into the eukaryotic DNA replication systems and provides a technical foundation for the robust sequencing of DNAs with an expanded genetic alphabet.
|
|
Scooped by
mhryu@live.com
Today, 7:14 PM
|
The KEGG Orthology (KO) system links DNA and protein sequences to biological functions and pathways, providing a curated, fundamental and consistent annotation framework across all domains of life. While accurate, traditional sequence alignment-based annotation methods are computationally expensive, which severely limits their application in large-scale datasets. To address this challenge, we introduce DeepKOALA, a deep learning approach based on Gated Recurrent Units (GRU), which frames KO annotation as an open-set recognition task. This design reduces false positives arising from out-of-scope sequences and, together with a lightweight GRU backbone, enables high-throughput annotation. The GRU-based model was benchmarked against four other deep learning architectures and showed the best balance between speed and accuracy. We further performed a cross-species evaluation of DeepKOALA in comparison with existing KO annotation tools, where it achieved an F1-score of 0.8653 and 37.5-fold acceleration compared with BlastKOALA. We also provide a specialized fragment model for handling incomplete sequences and an optional Multi-domain mode. Together, these features make DeepKOALA a scalable, efficient, and accurate solution for high-throughput function annotation.
|
|
Scooped by
mhryu@live.com
Today, 5:36 PM
|
Nitazenes are an emergent class of synthetic opioids that often rival or exceed fentanyl in their potency. These compounds have been detected internationally in illicit drugs and are the cause of increasing numbers of hospitalizations and overdoses. New analogs are consistently released, making detection challenging — new ways of testing a wide range of nitazenes and their metabolic products are urgently needed. Here, we develop a computational protocol to redesign the plant abscisic acid receptor PYR1 to bind diverse nitazenes and maintain its dynamic transduction mechanism. The best design has a low nanomolar limit of detection in vitro against nitazene and menitazene. Deep mutational scanning yielded sensors able to recognize a range of clinically relevant nitazenes and the common metabolic byproduct in a complex biological matrix with limited cross-specificity against unrelated opioids. Application of protein design tools on privileged receptors like PYR1 may yield general sensors for a wide range of applications in vitro and in vivo. Nitazenes are potent synthetic opioids that are difficult to detect. Here, authors computationally redesign a plant receptor to create sensitive sensors capable of detecting diverse nitazenes and their metabolites in biological samples.
|
|
Scooped by
mhryu@live.com
Today, 5:30 PM
|
CRISPR/Cas-based genome editing has revolutionized plant biotechnology, enabling precise genomic modifications for crop improvement and functional genomics. The success of these applications hinges on designing single guide RNAs (sgRNAs) that maximize on-target efficiency while minimizing off-target effects. Current resources for sgRNA design and performance evaluation in plants are fragmented and lack integration with genomic and epigenomic context, which influences both editing efficacy and specificity. Here, we present PCdb (Plant CRISPR Database; https://gmo.sjtu.edu.cn/pcdb), a comprehensive plant-focused database by integrating experimentally validated sgRNAs, their annotated genomic contexts, genome-wide off-target predictions, and multi-layered epigenomic annotations. PCdb encompasses 6,172 manually curated editing records from 2,132 publications, covering 4,320 unique sgRNAs and 6,117,424 predicted off-target sites across nine major plant species. Uniquely, PCdb contextualizes potential editing outcomes-both on-target and off-target-within the chromatin landscape by incorporating DNA methylation profiles, chromatin accessibility data, and histone modification patterns. The database features an intuitive web interface supporting flexible queries, interactive visualization tools, and comprehensive analytical modules for both sgRNA efficiency assessment and off-target analysis. A case study reanalysis of Oryza sativa yield-related genes demonstrates PCdb's capability to provide a comprehensive performance profile, evaluating both on-target characteristics and off-target risks within their native epigenomic context. Through systematic analysis of the database, we reveal critical sequence and chromatin features influencing editing outcomes, providing novel insights for improved gene editing efficacy and specificity.
|
|
Scooped by
mhryu@live.com
Today, 4:49 PM
|
Temperate phage can transmit both horizontally (lytic cycle) and vertically (lysogenic cycle). Many temperate phage have the ability to modify their lysis/lysogeny decisions based on various environmental cues. For instance, many prophage are known to reactivate when SOS stress responses of their host are triggered. Temperate phage infecting Bacilli can also use peptide signals (“arbitrium”) to control their lysis/lysogeny decisions. However, information from the arbitrium and SOS systems can be potentially conflicting, and it is unclear how phage integrate information carried by these two different signals when making lysis–lysogeny decisions. Here, we use evolutionary epidemiology theory to explore how phage could evolve to use both systems to modulate lysis/lysogeny decisions in a fluctuating environment. Our model predicts that it can be adaptive for phage to respond to both host SOS systems and arbitrium signaling, as they provide complementary information on the quality of the infected host and the availability of alternative hosts. Using the phage phi3T and its host Bacillus subtilis, we show that during lytic infection and as prophage, lysis–lysogeny decisions rely on the integration of information on host condition and arbitrium signal concentrations. For example, free-phage are more likely to lysogenise a stressed host, and prophage are less likely to abandon a stressed host, when high arbitrium concentrations suggest susceptible hosts are unavailable. These experimental results are consistent with our theoretical predictions and demonstrate that phage can evolve plastic life-history strategies to adjust their infection dynamics to account for both the within-host environment (host quality) and the external environment that exists outside of their host (availability of susceptible hosts in the population). More generally, our work yields a new theoretical framework to study the evolution of viral plasticity under the influence of multiple environmental cues.
|
|
Scooped by
mhryu@live.com
Today, 4:17 PM
|
Precise activation of endogenous genes is a powerful strategy for functional genomics and therapeutic development, but current CRISPR-based transcriptional activation (CRISPRa) systems are limited by the large size of Cas proteins for adeno-associated virus (AAV) delivery. Here, we present a high-efficiency dCas12f-based transcriptional activation system (HEAL), which recruits transactivators through MS2 coat protein binding to MS2 aptamers embedded within the sgRNA scaffold. Engineered to enhance DNA binding, nuclear localization, and transactivator recruitment, HEAL induces over 100,000-fold activation of endogenous genes and outperforms existing CRISPRa systems in vitro and in vivo. We further develop red-light-inducible OptoHEAL and small-molecule-inducible ChemHEAL for remote and precise transcriptional control. AAV-delivered HEAL targeting interleukin 10 alleviates acute kidney injury in mice, while ChemHEAL-mediated activation of thymic stromal lymphopoietin reduces body weight in obese mice. HEAL provides a modular, compact, and controllable platform for endogenous gene activation with strong potential for fundamental research and gene therapy. Precise activation of endogenous genes is a powerful strategy for research and therapy, but existing tools are too large for efficient delivery. Here, authors create a compact and programmable CRISPRa platform that can be remotely controlled and shows therapeutic benefits in mouse disease models.
|
|
Scooped by
mhryu@live.com
January 7, 11:50 PM
|
Aptamers are single-stranded oligonucleotides that bind molecular targets with high affinity and specificity, offering significant advantages over antibodies in therapeutic and diagnostic applications. However, their discovery through Systematic Evolution of Ligands by EXponential enrichment (SELEX) remains time-consuming, expensive, and susceptible to experimental biases. Here we present AptaBLE, a deep learning framework that combines pretrained protein and nucleic acid sequence encoders with a novel symmetric bidirectional cross-attention architecture to predict aptamer-protein binding interactions. This design enables robust generalization across diverse protein targets and ssDNA aptamer modalities while maintaining the ability to process variable-length sequences. We demonstrate that AptaBLE significantly outperforms existing methods, achieving superior accuracy in predicting aptamer-protein binding. Furthermore, we demonstrate two complementary de novo generation approaches that produce novel aptamers binding target proteins with desired specificity profiles and Kds as low as 31nM to-date. AptaBLE represents a significant advance in computational aptamer design, providing an accessible, sequence-based platform for accelerating therapeutic and diagnostic aptamer development.
|
|
Scooped by
mhryu@live.com
January 7, 11:07 PM
|
The ocean, Earth’s largest carbon reservoir, exerts a central role over atmospheric CO2 through its capacity to store carbon primarily as bicarbonate ions. Direct observations indicate that the global ocean has a net carbon uptake of 2.6–3.0 petagrams of carbon annually, representing nearly 30% of anthropogenic CO2 emissions. This review examines two principal domains of oceanic carbon cycling. The first concerns the natural uptake and storage of anthropogenic CO2, with emphasis on the response of the marine carbonate system and the spatial distribution of absorbed carbon. The second addresses emerging marine CO2 removal strategies, especially ocean alkalinity enhancement and macroalgae-based approaches. Ocean alkalinity enhancement aims to increase seawater buffering capacity to facilitate greater CO2 uptake, whereas macroalgae-based strategies rely on photosynthetic fixation and the subsequent storage of organic and inorganic carbon in various reservoirs. Effective implementation of these approaches necessitates rigorous monitoring, reporting, and verification frameworks to ensure their quantifiable efficacy and environmental integrity.
|
|
Scooped by
mhryu@live.com
January 7, 4:36 PM
|
Although genome sequencing technologies have advanced rapidly, microbial genomes still contain numerous genes with unknown functions, posing ongoing challenges for comprehensive genome annotation. Traditional annotation methods are constrained by a lack of scalable experimental techniques and the limitations of conventional homology-based computational approaches. Recent computational innovations, particularly deep learning, have substantially improved gene function prediction, facilitating more efficient annotation of transcription factors, enzymes and other protein classes. Integrating computational and experimental approaches has enabled the development of workflows that systematize gene function discovery, paving the way for faster, more accurate and comprehensive genome annotation. Continued refinement of these integrated methods holds great promise for deepening our understanding of microorganisms. Here we review recent advances in artificial intelligence for gene function discovery and discuss future directions for achieving interpretable and high-throughput artificial intelligence-guided annotation. In this Review, the authors discuss how artificial intelligence can aid microbial gene function discovery.
|
|
Scooped by
mhryu@live.com
January 7, 4:29 PM
|
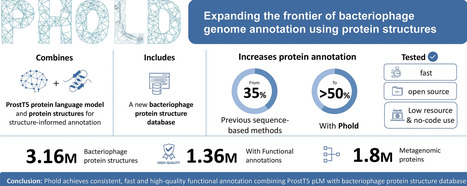
Bacteriophage (phage) genome annotation is essential for understanding their functional potential and suitability for use as therapeutic agents. Here, we introduce Phold, an annotation framework utilizing protein structural information that combines the ProstT5 protein language model and structural alignment tool Foldseek. Phold assigns annotations using a database of over 1.36 million predicted phage protein structures with high-quality functional labels. Benchmarking reveals that Phold outperforms existing sequence-based homology approaches in functional annotation sensitivity whilst maintaining speed, consistency, and scalability. Applying Phold to diverse cultured and metagenomic phage genomes shows it consistently annotates over 50% of genes on an average phage and 40% on an average archaeal virus. Comparisons of phage protein structures to other protein structures across the tree of life reveal that phage proteins commonly have structural homology to proteins shared across the tree of life, particularly those that have nucleic acid metabolism and enzymatic functions. Phold is available as free and open-source software at https://github.com/gbouras13/phold.
|
|
Scooped by
mhryu@live.com
January 7, 4:20 PM
|
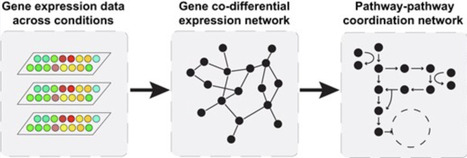
Gene co-expression networks are commonly used to identify functionally related genes, but they often suffer from spurious associations and fail to capture genes with restricted, context-specific responses. To address these limitations, we constructed YeastCoDEGNet, a global co-differential expression network in Saccharomyces cerevisiae, by reanalyzing microarray data from 143 carefully curated experiments comprising 425 comparisons. In this network, gene connections are defined by shared context-specific responses, enabling the identification of distinct topological groups enriched in either essential or nonessential genes, often associated with metabolic or nonmetabolic biological processes, respectively. We further characterized each gene by its responsiveness, essentiality, and number of co-differentially expressed partners, uncovering positional clustering of these features across chromosomal locations. To explore higher-order functional organization, we built a cross-pathway coordination network based on context-specific responses between pathway pairs, revealing modules of functionally related pathways. This network not only recovered well-known associations but also identified novel links, with particularly strong coordination observed among pathways involved in central carbon metabolism, amino acid and antioxidant processes, protein synthesis, trafficking, degradation, and gene regulation. By capturing context-specific gene expression dynamics, YeastCoDEGNet provides a powerful framework for studying how genes and pathways adapt to genetic and environmental perturbations.
|






























now NComm https://www.nature.com/articles/s41467-021-26791-x
Fibrin is a protein involved in the clotting cascade, which activates its polymerization to form blood clots. Fibrin’s polymerization is driven in part by noncovalent interactions between an alpha-chain domain present on the N-terminus of one fibrin monomer (i.e., the “knob” domain) and a gamma-chain domain on the C-terminus (i.e., the “hole” domain) of an adjacent monomer29. Our microbial ink design repurposes this binding interaction between alpha and gamma modules, i.e., the knob-hole interaction, to introduce non-covalent crosslinks between nanofibers and enhance mechanical robustness while maintaining shear-thinning properties