Your new post is loading...

|
Scooped by
?
Today, 1:28 PM
|
Over the next decade, mammalian synthetic biology should become the first precision control layer for human physiology. Synthetic gene circuits will sense molecules—glucose, lipids, and cytokines—and actuate closed-loop, real-time corrections integrated with host metabolism. This retires open-loop, Paracelsus-era dosing—pills at fixed intervals—in favor of continuous control. Placement should follow safety and access, not organ identity: subcutaneous sensing and control, by autologous cells, exosome/RNA cargoes, or xenogeneic tissue, and encapsulation or immune shielding manage risk. Metabolic control is modular—blood glucose does not need be controlled from the pancreas, nor do lipids need to be lowered by the liver. As circuits interface with metabolism, diagnosis, prevention, and therapy converge—shifting care from chronic management to cure.
|
|
Scooped by
?
Today, 1:01 PM
|
Vaccines are the most effective tool in preventing and managing infectious diseases. One of the critical challenges in vaccine development is the selection of suitable target antigens from the thousands of proteins produced by pathogens. Artificial intelligence is anticipated to play a significant role in addressing this challenge. In this study, we develop a framework termed PLGDL for protective antigen prediction that employs Protein Language and Geometric Deep Learning models. This framework leverages both primary sequence features and three-dimensional structural features of protein antigens, thereby reducing the biases associated with manually curated features. Our integrated model exhibits robustness across both constructed and public datasets and is applicable to viruses, bacteria, and eukaryotic pathogens. Notably, when applied to the ongoing Mpox outbreak, our model not only quickly identifies multiple known antigens but also discovers a protective antigen: G10R. Here, our study provides a high-performance screening tool for protective vaccine antigen prediction by synergistically utilizing the capabilities of protein language and geometric deep learning models, providing substantive insights and methodological advancements for rapid vaccine development. Vaccines are the most effective tool in managing infectious disease and characterizing features of protective epitopes could help in prediction methods. Here the authors use protein language and geometric deep learning frameworks to investigate primary sequence features and structural features to identify and predict potential antigens, showing prediction of a protective mpox epitope using this method.
|
|
Scooped by
?
Today, 12:29 PM
|
Heterogeneity within clonal cell populations remains a critical bottleneck within bioprocess engineering, notably by undermining bioproduction yields. Efforts to mitigate its impact have, however, been hampered by technological difficulties quantifying metabolism at the single-cell level. Here, we propose a framework based on single-cell biosensor analysis that enables robust characterization of cell’s metabolic states, leveraging it to detect and isolate isogeneic heterogeneity in response to environmental perturbations and within microbial cell factories. We identify acute and gradual glucose depletion to induce differentiation of metabolically distinct subpopulations and reveal these subpopulations to exhibit differential production capabilities, with lower intracellular pH subpopulations exhibiting enhanced product accumulation within violacein-producing strains but reduced yields within lycopene-producing strains. Lastly, we highlight galactose cultivation as a method to modulate subpopulation dynamics towards higher-producing lycopene phenotypes. Altogether, our research provides insights into subpopulation differentiation and establishes promising avenues for the engineering of more robust and higher-producing strains. Heterogeneity within clonal cell populations affects bioprocess engineering. Here, the authors report a biosensor-based toolkit to investigate phenotypic heterogeneity in engineered yeast, reveal pH-based subpopulations and metabolite production states, and modulate/shift subpopulation dynamics to increase lycopene production.
|
|
Scooped by
?
December 20, 11:56 PM
|
Bacteria exhibit two lifestyles: planktonic free-floating individual cells or sessile multicellular aggregates known as biofilms. The biofilm lifecycle is characterized by three distinct stages: attachment, maturation and dispersal. Specific signals govern each stage triggering responses that spatially and temporally regulate bacterial attachment to a surface, synthesis of extracellular matrix components and their subsequent degradation. Characterizing these signals is therefore a valuable approach to develop novel antibiofilm therapies. Here, we used the model biofilm-forming bacterium Pseudomonas aeruginosa PAO1 to characterize the transcriptional profiles of each stage of the biofilm life cycle: attachment, biofilm maturation and spontaneous dispersal. We report that surface attachment was accompanied by the upregulation of genes comprising the mechanosensor Pil-Chp, whereas biofilm maturation characterised with the upregulation of genes involved in Pel polysaccharide synthesis, siaD and PA4396 diguanylate cyclases as well as pipA, fimX and PA5442. In contrast, dispersing cells upregulated genes responsible for alginate, rhamnolipid, and extracellular nucleases (eddA, eddB) biosynthesis, as well as the transcriptional regulator of dispersal amrZ. Additionally, genes encoding the quorum sensing dispersal molecule cis-2-decenoic acid (dspS and dspI), canonical phosphodiesterases (nbdA and rbdA) and eleven other c-di-GMP–related enzymes were also upregulated during dispersal. Our comprehensive analysis of transcriptional profiles associated with different biofilm stages allowed us to define a subset of fourteen genes as biomarkers of biofilm dispersal. Our study therefore provides benchmarking stage-specific transcriptional profiles for P. aeruginosa biofilms in closed culture systems, which led to the identification of a dispersal fingerprint marking the onset of dispersal.
|
|
Scooped by
?
December 20, 11:33 PM
|
Richard Lenski traces the legacy of Escherichia coli and how science is evolving to use this model organism in new ways.
|
|
Scooped by
?
December 20, 11:30 PM
|
Monitoring acetic acid (AC) in fermentation processes is essential as excessive AC accumulation, particularly during alcoholic fermentation, can disrupt fermentation and lead to spoilage. However, conventional detection methods such as steam distillation, GC–MS, and HPLC are costly, time-consuming, and require liquid-phase samples, limiting their use for real-time monitoring and early identification of AC buildup. Here, we present an alternative tool for AC detection using a whole-cell bacterial biosensor, which utilizes the YwbIR transcriptional regulator from Bacillus subtilis. The designed biosensor exhibits high sensitivity, manifesting a linear response with (R2 = 0.97) from 0 to 1.0 g/L and a 5–8 fold induction at wine spoilage-relevant concentrations. It retains functionality in ethanol-rich matrices (up to 14.5% v/v) and enables headspace detection. Specificity assays and molecular docking analyses confirm high affinity for AC over other volatile fatty acids. This biosensor offers a low-cost solution for real-time AC monitoring, allowing timely intervention before spoilage occurs and supporting improved quality assurance in fermentation-driven food and beverage production.
|
|
Scooped by
?
December 20, 11:25 PM
|
Cell-surface receptors perceive environmental cues and trigger appropriate responses. In plants, these receptors comprise ectodomain, juxta-membrane, and cytosolic regions that define ligand specificity, modulate co-receptor associations, and fine-tune downstream signaling, respectively. Here we highlight the mechanistic principles underlying each module and discuss strategies to reprogram them. By integrating structural insights with illustrative examples, we provide a blueprint for designing cell-surface receptors with customized recognition specificity and programmable outputs, offering new opportunities to enhance plant resilience in the face of rapid climate change.
|
|
Scooped by
?
December 20, 10:45 PM
|
Point-of-use diagnostics based on allosteric transcription factors (aTFs) are promising tools for environmental monitoring and human health. However, biosensors relying on natural aTFs rarely exhibit the sensitivity and selectivity needed for real-world applications, and traditional directed evolution struggles to optimize multiple biosensor properties at once. To overcome these challenges, we develop a multi-objective, machine learning (ML)-guided cell-free gene expression workflow for engineering aTF-based biosensors. Our approach rapidly generates high-quality sequence-to-function data, which we transform into an augmented paired dataset to train an ML model using directional labels that capture how aTF mutations alter performance. We apply our workflow to engineer the aTF PbrR as a point-of-use diagnostic for lead contamination in water. We tune the sensitivity of PbrR to sense at the U.S. Environmental Protection Agency (EPA) action level for lead and modify the selectivity away from zinc, a common metal found in water supplies. Finally, we show that the engineered PbrR functions in freeze-dried cell-free reactions, enabling a diagnostic capable of detecting lead in drinking water down to ~5.7 ppb. Our ML-driven, multi-objective framework powered by directional tokens can generalize to other biosensors and proteins, accelerating the development of synthetic biology tools for biotechnology applications. Allosteric transcription factors (aTFs) are promising tools for environmental and human health monitoring. Here the authors develop a multi-objective, machine learning-guided method to engineer an aTF-based portable diagnostic for environment sensing of lead in drinking water at the legal limit.
|
|
Scooped by
?
December 20, 4:29 PM
|
The production of recombinant proteins in E. coli is often hampered by the formation of inclusion bodies. While fusion tags can enhance solubility, existing systems are hampered by a lack of standardization, with tags scattered across disparate plasmid backbones and inconsistent cloning sites, complicating parallel screening. To address this, we constructed a standardized series of expression vectors, termed pNX, by incorporating nine small fusion tags (SUMO, LD, ACP, BCCP, GB1, Fh8, SmbP, TolA, and TrxA) into a uniform pET-28b backbone. Each pNX vector features an identical configuration: a T7 promoter, an N-terminal fusion tag, a synthetic linker, a TEV protease cleavage site, and a multiple cloning site (MCS) flanked by dual 6×His tags. We evaluated this system using four model proteins (EcFabG, eGFP, XccXanA2, and XccXanL). Our results showed that specific tags significantly improved both the expression level and solubility of the target proteins without compromising their biological activity. Notably, the lipoyl domain (LD) was identified, to our knowledge for the first time, as an effective solubility enhancer. The standardized MCS enabled rapid, parallel cloning, facilitating the efficient screening of optimal fusion partners. The pNX vector series provides a versatile and efficient platform for enhancing the soluble expression of challenging recombinant proteins in E. coli, streamlining the empirical identification of ideal fusion tags.
|
|
Scooped by
?
December 20, 4:24 PM
|
The discovery of the antiviral-responsive RNA editing system is a significant advance in eukaryotic biology. It reveals new dimensions of RNA editing diversity and fungal antiviral strategies, and provides compelling evidence for the adaptive significance of RNA editing in host-virus coevolution. This discovery establishes a novel model for investigating both the molecular mechanisms of RNA editing and the evolutionary dynamics of altruistic defense.
|
|
Scooped by
?
December 20, 4:12 PM
|
Extracellular vesicles (EVs) are lipid-bound nanocarriers released by various eukaryotic cells and found in diverse bodily fluids. EVs have transitioned from being considered cellular waste disposers to significant players in intercellular communication and signaling. These EVs carry signature cargos of infected cells and thus can be helpful as biomarkers or prognostic markers for infectious diseases. Viruses can manipulate the EV biogenesis machinery in their own dissemination. EVs released from virus-infected cells can carry immune modulatory molecules, thus contributing to disease progression. This comprehensive review collates the information on the impact of EVs on viral infection and disease progression.
|
|
Scooped by
?
December 20, 4:00 PM
|
Antimicrobial resistance (AMR) presents a mounting global health crisis. Traditional antibiotic discovery methods are hindered by low throughput, frequent rediscovery of known compounds, and limited access to unculturable microbes and silent biosynthetic gene clusters (BGCs). Droplet microfluidics offers a transformative platform to overcome these barriers by enabling massively parallel, compartmentalized analysis at the single-cell level. In this perspective, we discuss how droplet microfluidics is reshaping the antibiotic discovery landscape by addressing long-standing limitations in cultivation, screening, and compound identification. We highlight recent advances in: (1) accessing microbial dark matter through high-throughput single-cell technologies, (2) activating cryptic BGCs and dissecting microbial interactions using microfluidic coculture platforms, (3) accelerating chemical dereplication and discovery of novel metabolites through integration with mass spectrometry-based tools, and (4) uncovering resistance and persistence mechanisms by linking antibiotic resistance genes to individual microbial hosts and single cell transcriptomes. Together, these innovations position droplet microfluidics as a powerful engine to accelerate antibiotic discovery and combat the global threat of AMR.
|
|
Scooped by
?
December 20, 3:53 PM
|
Biologically meaningful interpretation of transcriptomic datasets remains challenging, particularly when context-specific gene sets are either unavailable or too generic to capture the underlying biology. We here present InCURA, an integrative clustering strategy based on transcription factor (TF) motif occurrence patterns in gene promoters. InCURA takes as input lists of (i) all expressed genes, used solely to identify dataset-specific expressed TFs, and (ii) differentially regulated genes (DRGs) used for clustering. Promoter sequences of DRGs are scanned for TF binding motifs, and the resulting counts are compiled into a gene-by-TFBS matrix. InCURA then uses unsupervised clustering to infer gene modules with shared predicted regulatory input. Applying InCURA to diverse biological datasets, we uncovered functionally coherent gene modules revealing upstream regulators and regulatory programs that standard enrichment or co-expression analyses fail to detect. In summary, InCURA provides a user-friendly, regulation-centric tool for dissecting transcriptional responses, particularly in settings lacking context-specific gene sets.
|
|
|
Scooped by
?
Today, 1:07 PM
|
Antibody-based therapies have transformed the management of immune-mediated inflammatory diseases (IMIDs), but the need for frequent injections often leads to inadequate patient adherence and suboptimal long-term disease control. To address this challenge, we develop AIDEN (aid for IMIDs: engineered EcN), an engineered probiotic platform that enables oral delivery of therapeutic antibodies using synthetic biology. In this study, we assess the efficacy of AIDEN-IL17, a variant designed to secrete single-chain variable fragments targeting interleukin-17A (IL-17A), in murine models of psoriasis and inflammatory bowel disease. AIDEN-IL17 exhibits stable gut colonization and sustained in situ antibody production, resulting in moderate reduction of systemic IL-17A levels and significant amelioration of disease symptoms. Notably, the AIDEN platform is modular and adaptable for delivering a broad range of antibody therapeutics, offering a promising, patient-friendly strategy for the treatment of IMIDs.
|
|
Scooped by
?
Today, 12:54 PM
|
Cell-to-cell variability often limits the efficiency of microbial bioproduction, yet how individual cells fluctuate over time and how these fluctuations shape population-level output remain unclear. To address this issue, we tracked a heterologous betaxanthin pathway in Escherichia coli using microfluidics-assisted time-lapse microscopy, allowing simultaneous measurement of fluctuations in betaxanthin, its biosynthetic enzyme DOD and growth across generations. Here we show that over 50% of high betaxanthin producers become medium or low producers after two divisions. Betaxanthin variation primarily originates from DOD noise, with a smaller contribution from growth rate fluctuations. We further develop a stochastic model to explore various control circuits and find that pathway enzyme or metabolite-based growth selection strategies are most effective in enhancing production. We experimentally validate the model by coupling enzyme expression to nutrient availability, which enriches high producers and boosts titer by 4.4-fold. Our results highlight key sources of metabolic heterogeneity and provide a framework for designing robust microbial processes. Cell-to-cell variability limits efficient microbial production. Here, the authors track single cells to reveal enzyme noise as the main source of bioproduction variation, and by coupling growth to pathway performance, they selectively enrich high producers and substantially boost overall titres.
|
|
Scooped by
?
Today, 11:19 AM
|
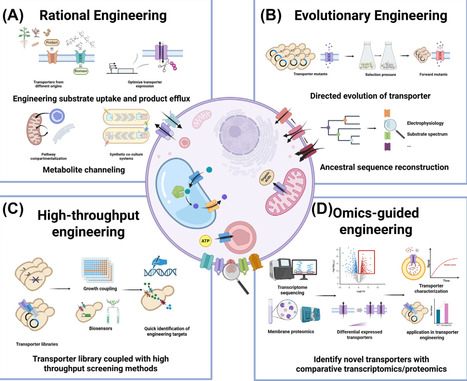
Membrane transporters play crucial roles in metabolite exchange, cellular communication, and metabolic homeostasis and are attractive targets in metabolic engineering for the development of microbial cell factories. While transporter engineering has proven effective in enhancing nutrient uptake, improving product secretion, and optimizing metabolic flux, its broader application is limited by incomplete knowledge of membrane transport systems and the sometimes high promiscuity of transporters, often resulting in unpredictable outcomes. This review provides an overview of recent progress in transporter engineering and characterization methods, highlighting their potential to enhance the production of bio-based chemicals.
|
|
Scooped by
?
December 20, 11:46 PM
|
Synthetic microbial communities (SynComs) are microbial consortia with defined taxonomic and functional traits, so that the combination elicits a predictable response under defined conditions. SynComs are artificially designed to enable inter-species metabolic interactions, metabolic division of labor, and ecological interactions that can elicit phenotypes like colonization stability and environmental adaptation. As an applied tool, SynComs have been deployed in diverse contexts, including agriculture, industry, and environmental ecology. This systematic review explores the processes used to construct SynComs, the mechanisms of metabolic interaction between members, and a review of the different ways that SynComs have been applied. We also explore the challenges for SynCom development and application, and future research directions that could overcome these challenges. SynComs are a powerful tool in our arsenal of applied technologies, but research and application are still nascent. While advances have been made, more research is needed to ensure SynCom technologies do not threaten global ecological security. SynCom technology represents a versatile platform for the controlled manipulation of microbial systems, enabling targeted modification of ecological and physiological processes. This emerging field marks a transition from descriptive biology toward a predictive and engineering-driven framework for understanding and shaping living systems.
|
|
Scooped by
?
December 20, 11:33 PM
|
Light-sensitive proteins allow organisms to perceive and respond to their environment, and have diversified over billions of years. Among these, Light-Oxygen-Voltage (LOV) domains are widespread photosensors that control diverse physiological processes and are increasingly used in optogenetics. Yet, the evolutionary constraints that shaped their protein dynamics and thereby their functional diversity remain poorly resolved. Here we systematically characterize the dynamics of 21 natural LOV core domains, significantly extending the spectroscopically resolved catalog through the addition of 18 previously unstudied variants. Using time-resolved spectroscopy, we uncover an exceptional kinetic diversity spanning from picoseconds to days and identify distinct functional clusters within the LOV family. These clusters reflect evolutionary branching, including a divergence of 1.0 billion years between investigated LOV variants from plants and 0.4 billion years of separation within one of these functional clusters. Individual variants with extreme photocycles emerge as promising anchor points for optogenetic applications, ranging from highly efficient adduct formation to ultrafast recovery. Beyond natural diversity, we introduce a LOV domain generated by artificial intelligence-guided protein design. Despite being sequentially remote from its maternal template, this variant retains core photocycle function while exhibiting unique biophysical properties, thereby occupying a new region on the biophysical landscape. Our work emphasizes how billions of years of evolution defined LOV protein dynamics, and how protein design can expand this repertoire, engineering next-generation optogenetic tools.
|
|
Scooped by
?
December 20, 11:26 PM
|
The gut microbiome plays a crucial role in maintaining health by supporting digestion, immunity, and overall well-being. Disruptions to the gut microbiome can result in dysbiosis, which is correlated with disease states. Recent advances in engineering the gut microbiome, functional ingredients designed through prebiotics, probiotics, and synbiotics have progressed together with synthetic microbial communities (SynComs), which influence the modulation of microbiome composition and functional role, offering a promising strategy to restore balance and enhance health. This field is rapidly advancing with broad applications focused on improving animal and human health. This review explores the significance and current applications of the engineering microbiome and its impact on gut health, as well as the challenges and sustainable future.
|
|
Scooped by
?
December 20, 10:49 PM
|
Although virus ecogenomics has expanded access to and understanding of the virosphere, existing classification tools lack taxonomic resolution and are unable to scale to modern discovery-based datasets or classify previously unknown sequence space. Here we develop vConTACT3—a machine learning-based tool that improves scalability and accuracy of virus taxonomy. By optimizing gene-sharing thresholds and leveraging adaptive, realm-specific cut-offs, vConTACT3 expands classification to both eukaryote and prokaryote viruses for four of the six officially recognized realms, and establishes accurate hierarchical taxonomy from genus to order. Specifically, vConTACT3 achieves >95% agreement with official taxonomy for 35,545 and 13,524 public prokaryotic and eukaryotic virus genomes, respectively, to surpass vConTACT2 across most realms, while still uniquely classifying previously uncharacterized taxa, and doing so even faster. vConTACT3 application provides taxonomy assignments for tens of thousands of unclassified taxa rapidly, automatically and systematically; evaluates virus sequence space to reveal support for fewer taxonomic ranks than currently available and identifies taxonomically challenging areas across the virosphere. vConTACT3 enables multirank, large-scale classification of eukaryotic and prokaryotic viruses.
|
|
Scooped by
?
December 20, 4:31 PM
|
Predicting changes in protein thermostability caused by amino acid substitutions is essential for understanding human diseases and engineering proteins for practical applications. While recent protein generative models demonstrate impressive zero-shot performance in predicting various protein properties without task-specific training, their strong unsupervised prediction ability remains underexploited to improve protein stability prediction. We present SPURS, a deep learning framework that rewires and integrates two complementary protein generative models–a protein language model and an inverse folding model–and reprograms this unified framework for stability prediction through supervised fine-tuning on mega-scale thermostability data. SPURS delivers accurate, efficient, and scalable stability predictions and generalizes to unseen proteins and mutations. Beyond stability prediction, SPURS enables broad applications in protein informatics, including zero-shot identification of functional residues, improved low-N protein fitness prediction, and systematic dissection of stability-pathogenicity for human diseases. Together, these capabilities establish SPURS as a versatile tool for advancing protein stability prediction and protein engineering at scale. Understanding how mutations alter protein stability is essential for biology and disease research. Here, the authors develop SPURS, a model that rewires pre-trained protein generative models to accurately predict stability changes.
|
|
Scooped by
?
December 20, 4:26 PM
|
Tumor-targeted bacteria have emerged as promising drug carriers due to their intrinsic motility and hypoxia-homing property. Therapeutic agents can be loaded onto the bacterial surface, enabling their active delivery into tumor tissues. However, premature drug release during systemic circulation—likely triggered by various physiological/physical factors—inevitably results in reduced efficacy or increased off-target toxicity. Here, we present a genetic engineering strategy that enables E. coli MG1655 (EC) to autonomously produce a biofilm “jacket” on its surface (termed MEC) by regulating the expression of the biofilm-associated Csg gene cluster. This biofilm coating markedly enhances drug adsorption (1.7-fold increase for the model drug indocyanine green, ICG) and effectively prevents off-target leakage during systemic circulation. Benefiting from its tumor-homing capability and biofilm-mediated protection, MEC can deliver substantially more ICG into tumor inner regions. In murine tumor models, MEC-mediated delivery achieves significantly enhanced intratumoral drug retention and photothermal efficacy in comparison with the wild-type bacterial carrier. This work demonstrates an effective tumor-targeted drug delivery strategy based on genetically engineered biofilm technology, offering a promising avenue for precision bacterial oncology.
|
|
Scooped by
?
December 20, 4:15 PM
|
Heme, a protoporphyrin IX iron complex, functions as an essential prosthetic group in hemoglobin and myoglobin, mediating oxygen storage and transport. Additionally, heme serves as a critical cofactor in various enzymes such as cytochrome c, enabling electron transfer within the mitochondrial respiratory chain. Unlike protein-bound heme, free or labile heme exhibits cytotoxic, pro-oxidant, and pro-inflammatory properties. Elevated levels of free heme are associated with various pathophysiological conditions, including hemolytic disorders such as sickle cell disease, malaria, and sepsis. In this review, we introduce the physiological roles of heme and its involvement in human health and disease. We also examine the mechanisms of heme sensing and regulation in bacterial cells. A variety of analytical methods have been developed to detect and quantify heme, enabling differentiation between protein-bound and free forms. These tools are discussed in the context of their applications in studying cellular heme regulation and their use in monitoring pathological conditions in humans. In particular, we describe examples of biosensors employing bacterial heme sensor proteins as recognition elements.
|
|
Scooped by
?
December 20, 4:10 PM
|
Bacteriophages are major drivers of bacterial population dynamics, yet the significance of post-transcriptional regulation during infection remains largely unexplored. Central to this regulatory layer are small RNAs (sRNAs), which regulate target mRNAs via base-pairing, typically facilitated by RNA chaperones such as Hfq. Here, we applied RNA interaction by ligation and sequencing (RIL-seq) to comprehensively map the in vivo RNA-RNA interaction network in E. coli during phage lambda infection. This analysis revealed extensive reprogramming of E. coli-E. coli interactions, phage-specific lambda-lambda interactions, and interkingdom interactions between phage and host RNAs. Among these, we identified a phage-encoded sRNA, phage replication enhancer sRNA (PreS), embedded within the early left operon. PreS regulates essential host genes, including dnaN, which encodes the DNA polymerase β sliding clamp. This regulation enhances DNA replication and fine-tunes the phage lytic cycle. These findings uncover an RNA-level regulatory layer in phage-host interactions and demonstrate how a phage-encoded sRNA can hijack host replication machinery to optimize infection.
|
|
Scooped by
?
December 20, 3:58 PM
|
CRISPR–Cas systems have revolutionized genome engineering technologies, but type IV CRISPR–Cas systems and their genome engineering potential have been critically underexplored. In this study, we identified a type IV-A3 CRISPR–Cas system from a clinical Klebsiella pneumoniae isolate and characterized its plasmid targeting activity and capacity to suppress chromosomal and plasmid gene expression in E. coli. We revealed the pivotal role of Csf3 (Cas5) and the dispensable roles of Csf1 (Cas8-like) and Csf4 (DinG helicase) subunits in IV-A3 CRISPR–Cas complex formation. The system prevents plasmid propagation via interplay between DinG helicase activity and strategic protospacer positioning relative to plasmid replication and maintenance components. We enabled the IV-A3 CRISPR–Cas system to introduce lethal, sequence-specific double-stranded (ds)DNA breaks in the E. coli chromosome by fusing the nuclease domain of the I-TevI nuclease to the Cas8 N-terminus. Further, we developed a series of base editors, with various editing efficiencies and windows, by fusing the PmCDA1 cytidine deaminase to the Cas8, Cas5, and DinG subunits. Finally, conjugative transfer of the Cas5–PmCDA1 base editor into E. coli deactivated the tryptophan repressor gene, boosting IAA production. Our study provides new insights into type IV-A3 CRISPR–Cas systems and highlights their potential in genome engineering applications.
|