 Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 3:56 PM
|
Genome engineering plays a crucial role in the rapidly growing fields of metabolic engineering and synthetic biology. Chromosomal integration and stable expression of functional genes or large metabolic pathways necessitate the development of host-independent enabling technologies in diverse bacteria. Here, a generalizable genome engineering approach, MNGE (Multi-targeting Non-specific Genome Engineering), is developed based on the multi-targeting integrase (MTI) systems for multi-copy (at least three copies), highly random (only requiring the core TT dinucleotide) integration of metabolic genes or pathways in both Gram-positive bacteria (Streptomyces and Saccharopolyspora) and Gram-negative bacteria (Burkholderia and Chromobacterium). Using MNGE, the fungicide UK-2 BGC (41 kb) and the polyether antibiotic salinomycin BGC (106 kb) were randomly integrated into a heterologous host Streptomyces albus, significantly enhancing their fermentation levels based on chromosome position effects. Furthermore, the potent Gq/11-signaling inhibitor FR900359 BGC (66 kb) was successfully expressed in Burkholderia gladioli by the MTI1 system. Together, the MNGE approach exhibits broad applicability for next-generation genome engineering in diverse bacteria, thereby achieving highly efficient production of high-value compounds.
|
|
Scooped by
mhryu@live.com
Today, 3:35 PM
|
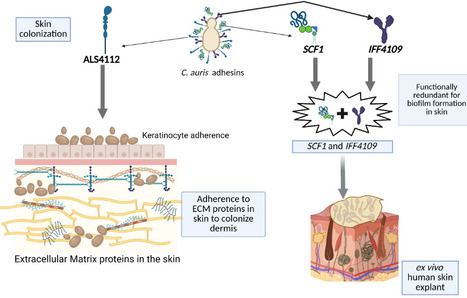
Candidozyma auris (formerly Candida auris) is an emerging multidrug-resistant fungal pathogen that causes life-threatening infections in humans. C. auris is distinct from other Candida species and exhibits exceptional capacity for skin colonization, resulting in nosocomial transmission and outbreaks of invasive infections. Fungal adhesins play a crucial role in skin colonization. With this perspective, we discuss the recent advances in the fungal adhesins of C. auris and how the divergence of adhesins in C. auris contributes to its unique fitness for skin colonization. We also discuss potential avenues to target fungal adhesins, which could pave the way for developing novel vaccine strategies and therapeutics to prevent skin colonization, nosocomial transmission, and invasive C. auris infections in humans.
|
|
Scooped by
mhryu@live.com
Today, 3:02 PM
|
Scientists say they have made some of the first direct measurements of how long it takes an individual, ordinary protein to fold. The results were surprising: they found no relationship between a protein’s sequence or size and how long it takes to fold into its 3D shape. And proteins seem to fold more efficiently than do other biomolecules, such as DNA — despite proteins having a more complex set of ingredients. The work was published today in Physical Review Letters. The authors attached a red dye molecule to one end of a string of amino acids, and a green one to the other end. The green dye shines on its own. The red dye is activated only when it receives energy from the green dye. Before the amino-acid string folds, the fluorescence from the green dye is visible. When the string starts folding, the two dye molecules are brought closer together, allowing energy to transfer from the green molecule to the red molecule, which then begins to shine. The fastest transition-path time was less than a microsecond, and the slowest was about four microseconds.
|
|
Scooped by
mhryu@live.com
Today, 2:54 PM
|
Developing short, stable, and potent antimicrobial peptides is a promising strategy to combat antibiotic resistance and persistence. We present CAMPER (Constraint-driven AMP Engineering with Ranking), a mechanistic artificial intelligence framework that integrates machine learning with biophysical ranking to prioritize membrane-targeting peptides effective against persister and biofilm forms of methicillin-resistant Staphylococcus aureus. We apply CAMPER to identify WP-CAMPER1 (12mer) that kills S. aureus MW2 at a minimal inhibitory concentration of 4 µg/mL. A 2% topical WP-CAMPER1 formulation reduces S. aureus MW2 burden by 2.5 log10 (p < 0.0002) in a murine prophylactic skin infection model, while its D-enantiomer, WP-CAMPER1-d, achieves 1.37 log10 (p < 0.0001) reduction in an established biofilm infection model. Single-cell analysis using a high-throughput microfluidic system shows that WP-CAMPER1-d reduces exponential-phase persisters of S. aureus USA300, and, in a deep-seated murine thigh infection model, decreases stationary-phase S. aureus MW2 persisters by 1.6 log10 (p < 0.0001). This study introduces CAMPER, a mechanistic artificial intelligence platform for designing antimicrobial peptides targeting MRSA. CAMPER identified a stable peptide that eradicates MRSA biofilms and persister cells and was active in mouse infection models.
|
|
Scooped by
mhryu@live.com
Today, 2:29 PM
|
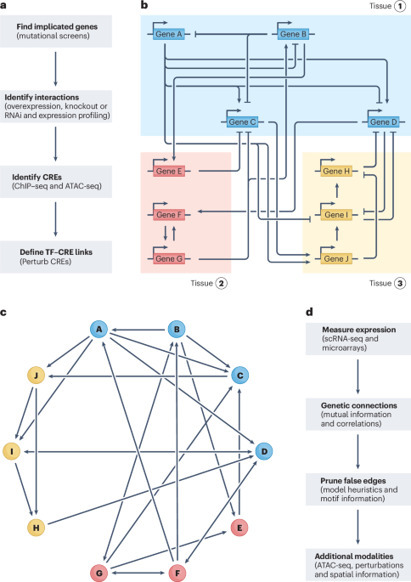
Gene regulatory networks (GRNs) explain how the genome controls cellular behavior and tissue morphogenesis, serving to connect molecular mechanism to functional output. Single-cell technologies now provide descriptions of these networks with unprecedented detail, but this advance has also revealed gene regulatory systems that are too complex for our existing conceptual frameworks. GRNs, which should provide mechanistic explanations, are increasingly reduced to statistical correlations — ‘hairballs’ that fail to capture molecular causation. Here, we explore why this dilemma exists and propose a path forward. We argue that methods in ‘representation learning’ can be used to model GRNs, without needing to capture every molecular detail. For this framework, we advocate three linked principles: models must be inherently mechanistic, with structures grounded in cellular and evolutionary biology; molecular principles and constraints must be used to reduce the solution space for learning GRN models; and more sophisticated forms of experimental perturbation and synthetic biological engineering are needed to train models and test predictions. By reimagining GRNs through these principles, we can bridge the gap from data abundance to new conceptual understanding. In this Perspective, Maizels and Briscoe discuss the limitations of current models of gene regulatory networks and outline solutions to harness data abundance without compromising explanatory power.
|
|
Scooped by
mhryu@live.com
Today, 12:54 PM
|
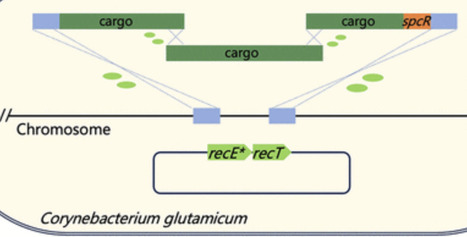
Corynebacterium glutamicum is a key industrial chassis for producing high-value chemicals, particularly amino acids. However, integration of large DNA fragments remains either inefficient or labor-intensive. Here, we optimized a RecET variant-assisted homologous recombination system to achieve single-step integration of DNA fragments larger than 11.3 kb by transforming donor DNA derived from either chromosome or linear DNA fragments. To further expand the size limit, we developed the One-Step Multi-Fragment Assembly Integration (OMAI) strategy, in which multiple overlapping PCR fragments are cotransformed and assembled in vivo, permitting integration of heterologous sequences with a length approximately 50% longer than the conventional single-fragment limit. Each editing cycle of OMAI is completed within 3 days, which is expected to be the most rapid method for large-fragment insertion in C. glutamicum.
|
|
Scooped by
mhryu@live.com
Today, 10:55 AM
|
Droplet digital (dd) CRISPR integrates the high sequence specificity of CRISPR-based nucleic acid detection with the absolute quantification capability of digital droplet microfluidics, offering high sensitivity, precision, and scalability. By partitioning samples into thousands to millions of picoliter microdroplets, ddCRISPR enables single-molecule resolution and minimizes background interference. This review summarizes the principles of droplet generation, manipulation, and detection in ddCRISPR platforms, as well as recent advances in amplification-based and amplification-free detection strategies. Representative applications are highlighted for viral, bacterial, and other DNA/RNA biomarker detection. Current challenges, including workflow automation, droplet stability, multiplexing, and assay portability, are discussed alongside future perspectives such as artificial intelligence (AI)-assisted analysis, point-of-care integration, and high-throughput multiplexed detection. These insights aim to guide the translation of ddCRISPR technologies from laboratory research to robust, scalable, and accessible diagnostic solutions.
|
|
Scooped by
mhryu@live.com
Today, 10:16 AM
|
Within lignocellulosic biomass, xylose is the second most abundant sugar after glucose. As a renewable and sustainable substrate, it is gaining attention as a feedstock for microbial bioprocesses. In this study, we demonstrated the co-production of polyhydroxybutyrate (PHB) and violacein from xylose. We initially confirmed the feasibility of co-production through genome-scale metabolic simulations, followed by optimization using a hybrid expression system that combines a conventional tac promoter and synthetic promoter-ribosome-binding site-terminator (semi-endo PRT) elements in dual plasmids. Additionally, we assessed the antimicrobial activity of violacein against type I methanotrophs. Recombinant E. coli DH5α harboring a hybrid system produced 111.3 ± 19.7 and 0.88 ± 0.23 mg/L of PHB and violacein, respectively, in M9 using xylose as the sole carbon source, without tryptophan supplementation. Using the synthetic PRT system, 528.9 ± 104 mg/g dry cell weight (DCW) of PHB was obtained. Additionally, violacein inhibits the growth of Methylomicrobium alcaliphilum 20Z at 9 µg/mL in nitrate mineral salt medium containing methanol as the sole carbon source. The use of lignocellulose-derived sugars for co-production offers an environmentally sustainable bio-manufacturing approach that contributes to greenhouse gas mitigation and supports the transition toward a circular bioeconomy.
|
|
Scooped by
mhryu@live.com
Today, 9:41 AM
|
The analysis of ribosome profiling (Ribo-Seq) data has provided evidence that many eukaryotic mRNAs contain translated upstream or downstream ORFs (uORFs/dORFs), but the biological significance of this translation activity remains, for the most part, unknown. One of the principal limitations has been the lack of Ribo-Seq data from several closely related species, precluding the identification of cases in which translation is phylogenetically conserved. Here, by combining Ribo-Seq data from 100 different experiments, we identify 2,332 translated uORFs and 1,008 translated dORFs in S. cerevisiae, which result in microproteins that tend to be highly hydrophobic or positively charged. To study their phylogenetic conservation, we have generated Nanopore direct RNA sequencing data, together with Ribo-Seq data, from six additional Saccharomyces species, spanning an evolutionary period of around 16 million years. We have identified 195 translated S. cerevisiae uORFs that are also translated in other Saccharomyces species; these uORFs are translated at levels comparable to the main coding sequence and display signatures of purifying selection at the level of the encoded microproteins. In contrast, dORFs are translated at very low levels and they are rarely conserved, suggesting much more limited microprotein functionalization. We have also discovered that uORF translation is associated with the formation of alternative transcript isoforms encompassing the region containing the uORFs but not the main protein coding sequence, implying that some microproteins can be produced independently of the main protein product. This work significantly advances our understanding of how initially pervasive uORF translation can result in new microproteins, providing many new candidates for further functional studies.
|
|
Scooped by
mhryu@live.com
March 8, 7:43 PM
|
Bacteria exhibit varying niche breadths, with generalists thriving in diverse environments and specialists confined to specific habitats. Although genes for cooperative traits have been suggested to influence niche breadth evolution, their precise role remains unclear. We used a combination of phylogeny-based comparative methods to test causal hypotheses about the directionality of the relationship between genes for cooperation and bacterial niche breadth evolution across 25,785 species. Our results revealed 1) a positive correlation between the proportion of genes for cooperation and niche breadth; 2) genes for cooperation influenced niche breadth evolution, with a decreased proportion of such genes promoting niche contraction as the predominant evolutionary direction; and 3) genes for cooperation experience more frequent gain and loss within species rather than across species. These findings suggest a role of bacterial cooperation in influencing niche breadth evolution and maintaining the ecological versatility of bacteria. While our results are consistent with a simple relationship under the hypotheses tested, more complex causal scenarios are possible, including the role of factors that influence both cooperation and niche breadth.
|
|
Scooped by
mhryu@live.com
March 8, 7:36 PM
|
Protein vesicles are spherical, hollow structures made entirely of folded proteins, fusion proteins, or polypeptides. Their intrinsic biocompatibility, nontoxicity, structural tunability, and cargo-loading capacity make them promising candidates for diverse biomedical applications. Although diverse forms of protein-based carriers have long been employed in drug, gene, and vaccine delivery, as well as in artificial antigen-presenting cells, vesicle architectures provide distinct advantages over free proteins, including enhanced stability, targeted delivery, and controlled release. We summarize recent advances in engineering protein vesicles and assess their current status within the broader landscape of synthetic vesicles in biomedicine. By comparing protein vesicles with liposomes, polymersomes, and virus-like particles, we highlight the limitations of conventional systems and underscore the unique benefits of protein-based assemblies. We further examine the emerging applications of protein vesicles in therapeutic delivery, diagnostics, and immunotherapy and discuss future directions needed to advance protein vesicle technologies toward clinical translation.
|
|
Scooped by
mhryu@live.com
March 8, 2:45 PM
|
Proteins are essential components of all living organisms and play a critical role in cellular survival. They have a broad range of applications, from clinical treatments to material engineering. This versatility has spurred the development of protein design, with amino acid sequence design being a crucial step in the process. Recent advancements in deep generative models have shown promise for protein sequence design. However, the scarcity of functional protein sequence data for certain types can hinder the training of these models, which often require large datasets. To address this challenge, we propose a hierarchical model named ProteinRG that can generate functional protein sequences using relatively small datasets. ProteinRG begins by generating a representation of a protein sequence, leveraging existing large protein sequence models, before producing a functional protein sequence. We have tested our model on various functional protein sequences and evaluated the results from three perspectives: multiple sequence alignment, t-SNE distribution analysis, and 3D structure prediction. The findings indicate that our generated protein sequences maintain both similarity to the original sequences and consistency with the desired functions. Moreover, our model demonstrates superior performance compared twith other generative models for protein sequence generation.
|
|
Scooped by
mhryu@live.com
March 8, 2:39 PM
|
Fungal biotechnology is crucial for generating high-value enzymes and fermentation products. Despite its industrial importance, major knowledge gaps in understanding fungal genomic variation, phenotypic diversity, and protein function prediction constrain biological innovation. While advancements in sequencing technologies have established data science as an integral component in driving developments in industrial fungal biotechnology, the inherent complexity of fungal genomes and incompatible repositories continue to limit comprehensive characterization of biological relationships and their translation into industrial applications. This review examines recent progress in non-graph methodologies applied to fungal biology. Genome annotation tools uncover genetic variation through homology-based approaches and enable functional annotation of sequence variants. Metric-based methods identify horizontal gene transfer events, while multivariate techniques characterize phenotypic variation across conditions. However, the increasing diversity, scale, and multimodal nature of fungal datasets require more integrative frameworks. Graph data science, a multivariate approach to model complex relationships as networks, offers opportunities to overcome these challenges. We discuss how graph-based methods enhance the detection of genomic structural variation and enable the modeling of molecular interactions. Furthermore, we outline how these approaches facilitate the exploration of complex fungal systems through multi-taxon, reference-free analyses, that integrate evolutionary signals, functional associations, and curated knowledgebases. By surveying available fungal resources and their taxonomic and ecological representations, we identify well-characterized genera, highlight underexplored taxa requiring further data generation, and pinpoint the ecological biases inherent in current sequencing efforts. Collectively, these advancements demonstrate how graph data science can accelerate fungal research and bridge fundamental discoveries and biotechnological applications.
|
|
|
Scooped by
mhryu@live.com
Today, 3:40 PM
|
Bacteria and fungi ubiquitously coexist, with their interactions critically influencing human health and industrial processes. Quorum sensing (QS) is a core regulatory mechanism that enables density-dependent coordination and phenotypic responses across these two kingdoms. While bacteria and fungi utilize their respective QS systems to engage in competitive or cooperative interactions to enhance their environmental adaptability, the current understanding of QS-based communications between them remains scattered, and a systematic summary of this field is still lacking. In this review, we examine the intricate dialog between bacteria and fungi, focusing on its role in microbial network assembly and ecosystem function, to provide a comprehensive analysis and engineering perspective on QS-based cross-kingdom communication. Specifically, we will first briefly delineate the core architecture of bacterial and fungal QS systems and the phenotypes they govern. Then, we will analyze QS-based interactions across diverse environments between different bacteria and fungi, categorizing natural QS interactions based on various phenotypes, including biofilm co-assembly and metabolic complementation. We further compare and analyze synthetic biology strategies, including promoter engineering and directed evolution of QS regulatory components, for reprogramming bacterial-fungal interactions and their applications. By synthesizing and contrasting these natural paradigms with synthetic designs, we provide a blueprint for achieving modular control over bacterial-fungal communities in diverse environments. Finally, by outlining persistent challenges and future trends, we aim to propel this field forward, enabling the deciphering of complex microbial interactions and ultimately increasing our capacity to engineer microbial consortia for diverse applications.
|
|
Scooped by
mhryu@live.com
Today, 3:24 PM
|
Microbial competition for trace metals shapes their communities and interactions with humans and plants. Many bacteria scavenge trace metals with metallophores, small molecules that chelate environmental metal ions. Metallophore production may be predicted by genome mining, where genomes are scanned for homologs of known biosynthetic gene clusters (BGC). However, accurately detecting non-ribosomal peptide (NRP) metallophore biosynthesis requires expert manual inspection, stymieing large-scale investigations. Here, we introduce automated identification of NRP metallophore BGCs through a comprehensive algorithm, implemented in antiSMASH, that detects chelator biosynthesis genes with 97% precision and 78% recall against manual curation. We showcase the utility of the detection algorithm by experimentally characterizing metallophores from several taxa. High-throughput NRP metallophore BGC detection enabled metallophore detection across 69,929 genomes spanning the bacterial kingdom. We predict that 25% of all bacterial non-ribosomal peptide synthetases NRPS encode metallophore production and that significant chemical diversity remains undiscovered. A reconstructed evolutionary history of NRP metallophores supports that some chelating groups may predate the Great Oxygenation Event. The inclusion of NRP metallophore detection in antiSMASH will aid non-expert researchers and continue to facilitate large-scale investigations into metallophore biology.
|
|
Scooped by
mhryu@live.com
Today, 2:56 PM
|
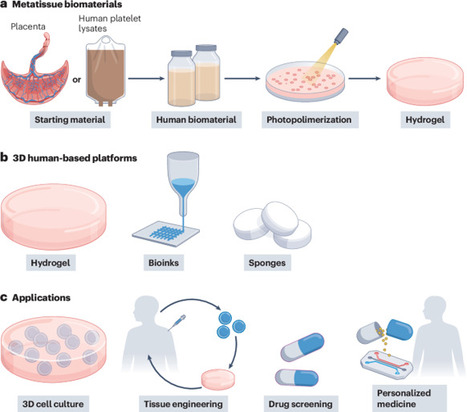
Human protein-based biomaterials facilitate the development of preclinical models that integrate predictive value and ethical responsibility. Metatissue uses decellularized extracellular matrix from placenta samples and human blood to create tunable, xeno-free substrates for 3D cell culture, minimizing the reliance on animal models and accelerating translational research.
|
|
Scooped by
mhryu@live.com
Today, 2:43 PM
|
Nucleic acid degradation is a common strategy for prokaryotic antiphage systems, as exemplified by the CRISPR-Cas system. The PD-(D/E)-XK nucleases constitute a widely distributed family in these defenses. Notably, most members exhibit a single nuclease domain, while variants containing dual nuclease domains within a single polypeptide remain underexplored, and their molecular mechanisms largely obscure. Here, we biochemically and functionally study a single-protein system containing an uncharacterized PD-(D/E)-XK defense protein (Upx). As revealed by single-particle electron cryo-microscopy (cryo-EM) structure, the C-terminal domain (CTD) harboring the conserved PD-(D/E)XK catalytic core is buttressed by the N-terminal domain (NTD) and the middle domain (MD). Functional assays demonstrate that the nucleic acid binding capability of the CTD is enhanced by the MD. The NTD also displays a noncanonical, basal exonuclease activity that is auto-inhibited by MD. IP-MS experiments identify Upx-interacting phage proteins, and substrate profiling defines its physiological preferences, collectively pointing to its potential physiological targets. Notably, the phage protein gp16 was found to relieve MD-mediated inhibition of the NTD, suggesting a virus-triggered mechanism for activating Upx’s dual nuclease activity. Together, these findings establish Upx as a single-protein dual-nuclease anti-phage system, expanding our understanding of bacterial immunity and informing antiviral strategy development. Bacteria employ diverse nucleases to defend against viruses. Here, authors characterize Upx, a single-protein, dual-nuclease system, and reveal that a specific phage protein relieves auto-inhibition to activate its antiviral function.
|
|
Scooped by
mhryu@live.com
Today, 12:58 PM
|
Adipic acid (AA) is an important dicarboxylic acid that serves as a precursor in the synthesis of nylon-6,6. Given the increasing market demand for AA and the environmental concerns associated with its conventional production, the development of sustainable biosynthetic techniques for AA production has become a key research focus in both industry and academia. However, industrially viable technologies for AA biosynthesis remain constrained by several challenges, particularly incomplete raw material utilization, low strain conversion efficiency, a complex fermentation process, and high costs of downstream separation. To overcome these barriers, this review presents the current state of AA biosynthesis, critically discussing biosynthetic pathways and advanced metabolic engineering strategies and tools for constructing cell factories with high conversion efficiency. The basic principles relevant to improving the fermentation process and downstream separation technologies are also comprehensively reviewed. Key challenges and knowledge gaps are identified, providing insights to guide future research toward commercially viable biobased AA production.
|
|
Scooped by
mhryu@live.com
Today, 11:01 AM
|
Biological research often involves complex, repetitive, and high-throughput manipulations that are well-suited to automation. However, current robotic systems generally excel only at narrowly defined tasks or standardized workflows and remain expensive, inflexible, and dependent on proprietary modules or reagents. To address these limitations, we developed the Open-Source Collaborative Automation & Robotics (OSCAR) platform, a flexible and low-cost system designed to perform common laboratory manipulations using standard, human-operated equipment. OSCAR incorporates open-source software and modular hardware to maximize accessibility and affordability. The platform features a robotic arm equipped with a dual-function end-effector: a pipetting module for precise liquid handling and a vision-enabled gripper for manipulating laboratory tools. To demonstrate the platform’s versatility, we implemented a representative plasmid assembly workflow, from PCR amplification and enzymatic assembly to transformation, plating, colony picking, PCR screening, and validation by agarose gel electrophoresis. By making this system open-source and compatible with widely used consumables and equipment, we aim to democratize access to automation and broaden its adoption across diverse research environments.
|
|
Scooped by
mhryu@live.com
Today, 10:36 AM
|
Insulin glargine, a long-acting insulin analog, is essential for diabetes treatment. However, its industrial production remains challenging due to limitations in conventional expression systems. Here, we employed the industrial filamentous fungus Trichoderma reesei as an alternative expression host for insulin glargine production, leveraging its superior protein secretion capacity. Initially, expression constructs containing the constitutive Pcdna1 promoter and CBH1 signal peptide (SP1) showed successful transcription but failed to achieve extracellular secretion, presumably due to induced endoplasmic reticulum (ER) stress. Consequently, we implemented a fusion protein strategy utilizing three distinct carrier proteins (CBH1, CBH2, and LA-20) to enhance glargine secretion. Notably, the CBH1 fusion not only enabled detectable glargine secretion but also significantly alleviated the ER stress. Furthermore, replacement of SP1 with the Aspergillus niger β-glucoamylase (glaA) signal peptide achieved a fourfold enhancement in glargine secretion and further reduced cellular stress responses. Following these systematic optimizations, a final yield of 58.95 mIU/L glargine was achieved in shake-flask cultures. Thus, the combined strategy described here could achieve extracellular production of glargine in T. reesei, suggesting that it is a promising host for secretory production of therapeutic recombinant proteins, particularly complex analogs like glargine.
|
|
Scooped by
mhryu@live.com
Today, 10:12 AM
|
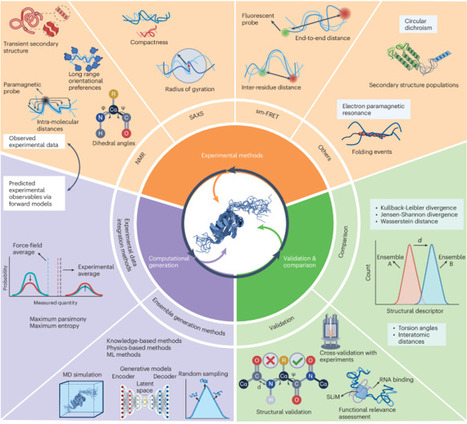
Disordered proteins play essential roles in myriad cellular processes, yet their structural characterization remains a major challenge due to their dynamic and heterogeneous nature. Here we present a community-driven initiative to address this problem by advocating a unified framework for determining conformational ensembles of disordered proteins. Our aim is to integrate state-of-the-art experimental techniques with advanced computational methods, including knowledge-based sampling, enhanced molecular dynamics and machine learning models. The modular framework comprises three interconnected components: experimental data acquisition, computational ensemble generation and validation. The systematic development of this framework will ensure the accurate and reproducible determination of conformational ensembles of disordered proteins. We highlight the open challenges necessary to achieve this goal, including force-field accuracy, efficient sampling, and environmental dependence, advocating for collaborative benchmarking and standardized protocols. This Perspective establishes a comprehensive and practical framework to guide intrinsically disordered protein (IDP) ensemble determination, benchmarking and interpretation, as well as proposes a roadmap for IDP ensemble determination, uncertainty quantification and actionable benchmarking strategies.
|
|
Scooped by
mhryu@live.com
Today, 9:30 AM
|
Aspergillus fumigatus is the leading cause of invasive mold infections in immunocompromised patients. Current antifungal treatment primarily depends on the triazole antifungals, which act by inhibiting Erg11/Cyp51, a key enzyme in the ergosterol biosynthetic pathway. However, resistance is emerging at an increasing rate, reducing treatment efficacy and patient survival. Confirmed resistance mechanisms in clinical isolates include mutations in cyp51A, cyp51B, hmg1, hapE, cox10, and the overexpression of drug efflux pumps. To identify additional determinants of triazole resistance, we grew A. fumigatus wild-type and Δcyp51A mutant strains under increasing concentrations of voriconazole. Sequencing of the resultant resistant strains identified known mutations in cyp51A and cyp51B, and novel mutations in hmg1, abcC (cdr1B), ptaB, erg25B, and srbA. Mutations of hmg1 and ptaB occurred early during evolution, while mutations of erg25B and srbA occurred later. Reintroduction of the novel mutations in hmg1, abcC, ptaB, and erg25B into wild-type A. fumigatus and correction of the srbA mutation in the evolved strain validated their contribution toward triazole resistance. Sterol profiling analysis indicated that mutation or deletion of erg25B is associated with a decrease in the accumulation of methylated sterols. Mutation or deletion of ptaB resulted in increased cyp51A, cyp51B, and erg25A expression. Sequence analysis of clinical isolates revealed enrichment of missense mutations in ptaB, hmg1, abcC, and srbA among triazole-resistant strains.
|
|
Scooped by
mhryu@live.com
March 8, 7:40 PM
|
All organisms fuel and build themselves through their energy metabolism. While classic biochemistry conceptualizes the fluxes of energy and matter, our understanding of how energy metabolism works in vivo contains major gaps. One reason is that energy metabolism spans multiple scales, from enzymes to organs, and shifts dynamically at environmental and developmental transitions, resulting in a degree of complexity that is presently impossible to capture. Genetically encoded fluorescent biosensors have started to bridge several critical gaps by enabling live monitoring of metabolites across scales. Recently, several paradigms of energy metabolism have started to shift, driven by the expansion of biosensing tools. This review explores advancements in our understanding of plant energy metabolism driven by fluorescent protein biosensing, highlights emerging concepts and open questions, and discusses how available tools, and much-needed future innovations, can unlock the potential of biosensing toward understanding in vivo plant energy metabolism and its effective modification.
|
|
Scooped by
mhryu@live.com
March 8, 7:33 PM
|
Therapeutic messenger RNAs (mRNAs) offer a versatile platform for treating a wide range of diseases, but their clinical efficacy hinges on optimizing both stability and translational efficiency. This review summarizes recent advances in strategies to enhance mRNA performance, with a focus on human therapeutics. We discuss secondary structure optimization, including artificial intelligence–guided design tools like RNAdegformer and LinearDesign, which balance structural stability and translational output. The roles of 5′ and 3′ untranslated regions in ribosome recruitment and mRNA decay are examined, highlighting sequence motifs and empirical design strategies that facilitate these processes. Chemical modifications, such as pseudouridine substitution, are shown to improve stability and reduce immunogenicity. Emerging approaches using circular RNA further extend transcript longevity. Finally, we review delivery technologies, including lipid nanoparticles, polymers, and extracellular vesicles, that protect mRNA and enable targeted cellular uptake. Together, these advances provide a road map for developing stable, efficient, and clinically viable mRNA-based therapeutics.
|
|
Scooped by
mhryu@live.com
March 8, 2:43 PM
|
Protein–protein interactions (PPIs) are central to cellular signaling and regulation, and their dysregulation underlies many diseases. Predicting the impact of mutations on PPI stability, quantified as ΔΔG, is essential for understanding disease mechanisms and guiding protein engineering. Here, we first present MutPPI, a graph-based deep-learning model that encodes full-residue structural features of protein–protein complexes and employs a shared GIN-GAT feature extractor for wild-type and mutant complexes. MutPPI outperforms 12 existing methods on an antibody–antigen single-point mutation dataset (S645). By integrating evolutionary information from protein language models, we further develop MutPPI-plus, achieving enhanced predictive performance. Second, we proposed a mutation-path-based data augmentation strategy, which enriches input modalities and improves generalization of both MutPPI and MutPPI-plus. After data augmentation, MutPPI-plus demonstrates state-of-the-art performance on S645 and three additional multi-point mutation datasets (SM_ZEMu, SM595, SM1124), substantially surpassing DDMut-PPI. Our analyses highlight the benefits of the multimodal framework and the physically informed data augmentation method. Together, these results provide a versatile computational tool for accurate ΔΔG prediction, advancing rational protein design.
|






























drop-seq (droplet-seq http://mccarrolllab.org/dropseq/) in plant roots.