Your new post is loading...
Your new post is loading...

|
Scooped by
Gilbert C FAURE
March 5, 2022 9:58 AM
|
Background: Shigella spp. and enterotoxigenic Escherichia coli (ETEC) cause high morbidity and mortality worldwide, yet no licensed vaccines are available to prevent corresponding infections. A live attenuated non-invasive Shigella vaccine strain lacking LPS O-antigen and expressing the ETEC...
|
|
Scooped by
Gilbert C FAURE
December 30, 2021 4:18 AM
|
Abstract The nose is the initial site of viral infection, replication, and transmission in the human body. Nasally inhaled vaccines may act as a promising alternative for COVID-19 management in add...
|
|
Scooped by
Gilbert C FAURE
December 30, 2021 4:11 AM
|
Vaccines in emergency use are efficacious against COVID-19, yet vaccine-induced prevention against nasal SARS-CoV-2 infection remains suboptimal.Since…...
|
|
Scooped by
Gilbert C FAURE
September 14, 2021 5:16 AM
|
Researchers have collaborated to resolve the aforementioned problems associated with injection vaccines to create a thermostable vaccine that can be taken orally.
|
|
Scooped by
Gilbert C FAURE
August 7, 2021 12:05 PM
|
Thai virologists at BIOTEC have 2 nasal spray options currently in development that may act as a substitute for Covid-19 vaccines in needle form.

|
Suggested by
LIGHTING
June 26, 2021 11:39 AM
|
Scientists use a protein found in mucus membranes to ferry vaccines to the lymph nodes.
|
|
Scooped by
Gilbert C FAURE
May 19, 2021 1:52 PM
|
New technologies are expected to enhance nasal vaccines industry growth as particular attention is being paid to designing delivery strategies that take into account the broad range of diseases, populations and healthcare delivery settings that will benefit from the mucosal route of delivery.
|
|
Scooped by
Gilbert C FAURE
March 27, 2021 3:32 AM
|
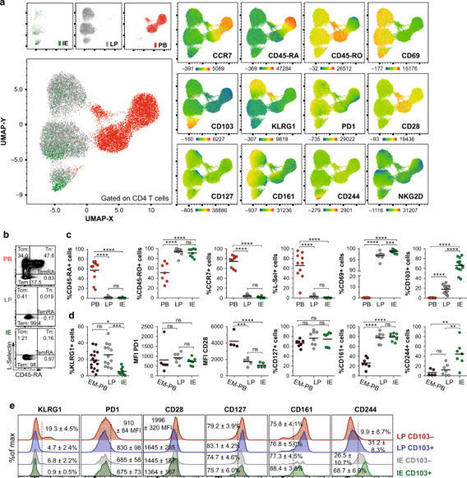
Studies in mice and humans have shown that CD8+ T cell immunosurveillance in non-lymphoid tissues is dominated by resident populations. Whether CD4+ T cells use the same strategies to survey peripheral tissues is less clear. Here, examining the turnover of CD4+ T cells in transplanted duodenum in humans, we demonstrate that the majority of CD4+ T cells were still donor-derived one year after transplantation. In contrast to memory CD4+ T cells in peripheral blood, intestinal CD4+ TRM cells expressed CD69 and CD161, but only a minor fraction expressed CD103. Functionally, intestinal CD4+ TRM cells were very potent cytokine producers; the vast majority being polyfunctional TH1 cells, whereas a minor fraction produced IL-17. Interestingly, a fraction of intestinal CD4+ T cells produced granzyme-B and perforin after activation. Together, we show that the intestinal CD4+ T-cell compartment is dominated by resident populations that survive for more than 1 year. This finding is of high relevance for the development of oral vaccines and therapies for diseases in the gut.
|
|
Scooped by
Gilbert C FAURE
February 6, 2021 9:58 AM
|
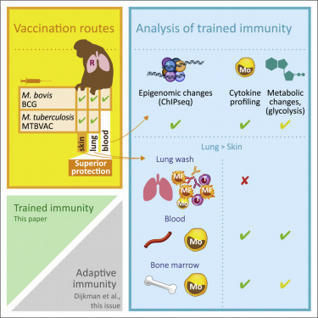
Vierboom et al. demonstrate the induction of trained immunity in blood and bone marrow
monocytes after vaccination with live attenuated TB vaccines in nonhuman primates. Mucosal respiratory delivery of BCG or MTBVAC induces trained immunity more efficiently compared to standard intradermal...
|
|
Scooped by
Gilbert C FAURE
January 9, 2021 4:20 AM
|
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) was first reported in late 2019 in China and is the causative agent of the coronavirus disease 2019 (COVID-19) pandemic. To mitigate the effects of the virus on public health, the economy and society, a vaccine is urgently needed. Here I review the development of vaccines against SARS-CoV-2. Development was initiated when the genetic sequence of the virus became available in early January 2020, and has moved at an unprecedented speed: a phase I trial started in March 2020 and there are currently more than 180 vaccines at various stages of development. Data from phase I and phase II trials are already available for several vaccine candidates, and many have moved into phase III trials. The data available so far suggest that effective and safe vaccines might become available within months, rather than years. The development of vaccines against SARS-CoV-2 is reviewed, including an overview of the development process, the different types of vaccine candidate, and data from animal studies as well as phase I and II clinical trials in humans.
|
|
Scooped by
Gilbert C FAURE
December 8, 2020 12:13 PM
|
Studies suggest that SARS-CoV-2-specific IgA antibodies are more neutralising and therefore COVID vaccines should encourage an IgA response.
|
|
Scooped by
Gilbert C FAURE
October 8, 2020 4:10 AM
|
KEY POINTS Pre-existing BCG immunity influences T cell responses of subunit booster vaccines. M. tuberculosis–specific subunit vaccines bypass this mechanism and improve protection. Abstract Despite the fact that the majority of people in tuberculosis (TB)–endemic areas are vaccinated with the Bacillus Calmette–Guérin (BCG) vaccine, TB remains the leading infectious cause of death. Data from both animal models and humans show that BCG and subunit vaccines induce T cells of different phenotypes, and little is known about how BCG priming influences subsequent booster vaccines. To test this, we designed a novel Mycobacterium tuberculosis–specific (or “non-BCG”) subunit vaccine with protective efficacy in both mice and guinea pigs and compared it to a known BCG boosting vaccine. In naive mice, this M. tuberculosis–specific vaccine induced similar protection compared with the BCG boosting vaccine. However, in BCG-primed animals, only the M. tuberculosis–specific vaccine added significantly to the BCG-induced protection. This correlated with the priming of T cells with a lower degree of differentiation and improved lung-homing capacity. These results have implications for TB vaccine design. This article is featured in Top Reads, p.1979 Introduction Mycobacterium tuberculosis infection is the leading cause of death because of a single infectious agent and has been in the top 10 causes of death worldwide for years (1).The only vaccine currently available against tuberculosis (TB) is M. bovis Bacillus Calmette–Guérin (BCG). When administered in early life, BCG efficiently prevents severe forms of childhood TB, but the efficacy against pulmonary disease in adulthood, the most common form of TB disease, is variable (2, 3). Thus, a new vaccine that prevents active pulmonary TB is needed to reduce M. tuberculosis transmission and TB-related mortality. CD4 T cells have been shown to be critical for host resistance to M. tuberculosis infections and are therefore the most common cell type targeted in preclinical and clinical TB vaccine development (4, 5). During M. tuberculosis infection, Ag expression and presentation have a major effect on differentiation and function of CD4 T cells. Recent mouse studies have shown that TB infection drives the differentiation of M. tuberculosis–specific CD4 T cells away from central memory T cells (e.g., secreting IL-2) toward effector/effector memory T cells that predominantly secrete IFN-γ (6). This results in a loss of self-renewing T cell subsets because of an impairment in the IL-2–producing capacity and a reduced capacity to traffic into the infected lung parenchyma (7, 8). Circulating T cells’ ability to populate the lung parenchyma has been established as a necessity for T cell–mediated protection in the lung (7, 9), possibly because a direct recognition of infected cells by CD4 T cells via the TCR is required (10). Thus, the ability to resist functional differentiation and home into inflamed lung tissues are key features for long-term protective CD4 T cells (7, 9). Similar to M. tuberculosis infection, the live mycobacterial BCG vaccine has been shown to promote differentiation and functional exhaustion of CD4 T cells in parenteral BCG-vaccinated mice, resulting in a failure to efficiently maintain long-term protection against M. tuberculosis (11–13). A recent study comparing different TB vaccines in clinical testing suggests that BCG may also induce more differentiated T cells than subunit vaccines in people (14). In line with this, we have recently shown that vaccination with an adjuvanted protein subunit vaccine (H56/CAF01) elicits less differentiated CD4 T cells with the capacity to localize to the infected parenchyma (15). In naive animals, such T cells are readily induced, but it has proven difficult to substantially reprogram the immune response after M. tuberculosis exposure by subunit vaccination (16–20). Given the similarities between the immune response arising from BCG vaccination and M. tuberculosis infection, we hypothesize that pre-existing “BCG-imprinted” T cells dictate the phenotype of the immune response induced by subsequent subunit booster vaccines. To investigate this, we designed two novel vaccines, H64 and H74, that selectively incorporated M. tuberculosis–specific (or “non-BCG”) Ags and compared the protective efficacy and T cell phenotype to a known booster vaccine sharing all of its Ags with BCG (H65) (21). Thus, H65 was tested as a classical BCG booster vaccine, whereas the H64 and H74 subunit vaccines supplement BCG’s Ag repertoire with M. tuberculosis–specific Ags. For comparability, the three vaccines consisted of six Ags that were either secreted by or associated with the type VII secretion system (also called the ESX secretion systems). H64 and H74 consisted of ESX-1–associated Ags (M. tuberculosis specific), whereas H65 consisted of Ags associated to ESX-2, 3, and 5 (also present in BCG). We first confirmed that the ESX-1 Ags were protective in mice and guinea pigs and that the level of protection was similar to the BCG boosting vaccine H65. We then moved on to show that in BCG-primed mice, the CD4 T cells induced by the H65 booster vaccine were more differentiated than the CD4 T cells induced by the ESX-1–based vaccine. Importantly, the T cells specific for the ESX-1 vaccine maintained their polyfunctionality and low differentiation status during chronic M. tuberculosis infection and were superior in entering the M. tuberculosis–infected lung parenchyma. As a result, the lung bacterial burden was significantly decreased in the BCG plus ESX-1 vaccination group compared with the groups with either BCG alone or BCG boosted with the H65 vaccine. These data add to the body of evidence supporting the use of ESX-1–associated (or other M. tuberculosis–specific/non-BCG) Ags in future TB vaccines (16, 22–25) and address the potential influence of BCG priming on subsequent booster vaccines. Materials and Methods Animals Six- to ten-week-old female mice or 400–500 g outbred female Hartley guinea pigs (Charles River Laboratories) were rested for 1 wk prior to initiation of any experimental procedures. Except for the M. tuberculosis Beijing HN878 challenge study, all mouse experiments were performed with female CB6F1 mice (Envigo) at Statens Serum Institut according to the Danish Ministry of Justice and Animal Protection Committees under permit 2014-15-2934-01065 and in compliance with European Union Directive 2010/63/EU. Mice were provided with radiation-sterilized food (Harlan Scandinavia) and water ad libitum and handled in accordance with the Danish Ministry of Justice and Animal Protection Committee regulations by authorized personnel. Infected mice were housed in a biosafety level 3 facility in cages contained within laminar flow safety enclosures (Scantainer, Scanbur). In the challenge study with the hypervirulent M. tuberculosis Beijing HN878, female C57BL/6 mice were purchased from SLC (Shizuoka, Japan). All animal experiments were performed according to the Korean Food and Drug Administration regulations and guidelines. The experimental protocols were reviewed and approved by the Ethics Committee and Institutional Animal Care and Use Committee (Permit Number: 2017-0264). All in vivo experiments were carried out under barrier conditions in an animal biological safety level 3 facility at the Avison Biomedical Research Center at Yonsei College of Medicine. Guinea pigs were maintained under animal biosafety level 3 barrier conditions in isolator cages (Thoren Caging Systems, Hazleton, PA) at Colorado State University. All experimental procedures were conducted in accordance with the Public Health Service Policy on the Humane Care and Use of Laboratory Animals and approved by the Colorado State University Institutional Animal Care and Use Committee (approval no. 13-4565A). Recombinant proteins All DNA constructs used in this study were made by chemical synthesis and codon optimized for expression in Escherichia coli before insertion into the pJ 411 expression vector (ATUM, Menlo Park, CA). Hybrid H64 and H74 were protein fusions without linkers between the six open reading frames. To minimize protein aggregation, all codons encoding cysteine were replaced with serine codons, five in H74 and three in H64. In both fusions, we added a His tag at the N-terminal end (MHHHHHH-). After transformation into E. coli BL21 (DE3) (Agilent Technologies), protein expression was induced with 1 mM isopropyl β-d-1-thiogalactopyranoside in 3-l cultures, and the proteins were purified from inclusion bodies by a three-step process as previously described (26). Hybrid H56 and H65 were designed as earlier described (21, 27) and expressed and purified in the same way as H74 and H64. The products were pure full-length products (>99% purity) with a protein concentration between 0.3 and 0.7 mg/ml and a total yield between 4 and 15 mg for the protein batches produced for this work. The identity of all purified protein batches was confirmed by mass spectrometry analysis (matrix-assisted laser desorption/ionization–time-of-flight). Immunizations and infections Mice were immunized s.c. three times at 2-wk intervals at the base of the tail with the fusion protein formulated in a cationic liposome adjuvant. Cationic liposomes (CAF01, 250 μg dimethyldioctadecyl-ammonium/50 μg trehalose 6,6-dibehenate) were emulsified with 1–10 μg fusion protein in 10 mM sterile Tris buffer (pH 7.4) to a final volume of 200 μl for each injection. Negative control mice received three equivalent doses of saline, and positive control mice received a single dose of 1 × 105 CFU M. bovis BCG Danish 1331 (Statens Serum Institut, Copenhagen, Denmark) given s.c. in the first round of immunization. In M. bovis BCG boost experiments, mice received one s.c. injection of 1 × 105 CFU M. bovis BCG Danish 1331 and were rested 8–26 wk, depending on the experiment, before vaccination three times with fusion protein as described above. Six weeks after the third immunization, mice were challenged with 50–100 M. tuberculosis strain Erdman (American Type Culture Collection), H37Rv (American Type Culture Collection 27294), Kazakhstan (mycobacterial interspersed repetitive units [MIRU] 1270-52), Vietnam (MIRU 1393-252), Beijing (MIRU 94-32), or Beijing HN878 suspended in PBS Tween 20 (0.05%). For M. tuberculosis strain Erdman, H37Rv, Beijing, Vietnam, and Kazakhstan, performed at Statens Serum Institut, we used a Biaera exposure system controlled by the AeroMP aerosol management, and for the hypervirulent M. tuberculosis strain Beijing HN878 experiment, performed at Yonsei College of Medicine, mice were infected with 60–70 virulent mycobacteria per mouse via the respiratory route using the inhalation chamber (Glas-Col, Terre Haute, IN). Guinea pigs (10/group), housed at Colorado State University, were immunized via the i.m. route and rested for 10 wk. A saline-treated and an intradermal M. bovis BCG–vaccinated group (inoculated with 103 CFU via the intradermal route) were included as a negative and positive control, respectively. Ten weeks postvaccination, guinea pigs were infected with a low-dose aerosol delivering ∼10 viable mycobacteria of virulent M. tuberculosis strain H37Rv into the lung of each animal. The animals were euthanized when they reached the set criteria established by the Institutional Animal Care and Use Committee, such as being moribund or exceeding acceptable weight loss and/or being affected in their respiratory rate (labored/heavy breathing). The body temperature was measured to track the clinical progression of the disease. For this, guinea pigs received a s.c. microchip implant (IPT-300 Bio Medic Data Systems, Seaford, DE) that allowed for the measurement of temperature and also carried information about experiment number and animal number. The body temperatures of individual guinea pigs were assessed each day at approximately the same time in the afternoon using a DAS-6006/7 scanner transponder (Bio Medic Data Systems). Isolation of cells and CFU measurements Spleen and lymph nodes from individual animals were kept at 4°C until processed through 70-μm nylon cell strainers (BD Pharmingen) followed by two washes and resuspension of the mononuclear cells in RPMI 1640 containing 5% FBS. Isolated lungs were transferred into Miltenyi C tubes containing HEPES/RPMI 1640 supplemented with collagenase (Roche/Sigma). The lungs were subsequently homogenized and digested for 30–45 min at 37°C and passed through cell strainers (BD Biosciences). After washing, the cells were resuspended in RPMI 1640 containing 5% FBS and stored at 4°C until use. For CFU measurements, lung homogenates were prepared in PBS Tween 80 (0.05%) from individual mice and plated at 3-fold serial dilutions on Middlebrook 7H11 Bacto Agar. After 3 wk of incubation at 37°C, the CFU were enumerated. Flow cytometry Single-cell suspensions of splenocytes or lung mononuclear cells (2 × 106 cells/well) were stimulated in vitro in V-bottom 96-well plates at 37°C in 200 μl complete media containing anti-CD49d (1 μg/ml) and anti-CD28 (1 μg/ml) Abs in the presence of rAg (2 μg/ml) for 1 h. Subsequently, 10 μg/ml brefeldin A (Sigma-Aldrich) was added, and the incubation continued for another 5–6 h. Following overnight storage at 4°C, cells were washed in FACS buffer (PBS containing 0.1% sodium azide and 1% FBS) and stained 30 min at 4°C for surface markers with mAbs as indicated. We used 1:400 dilutions of anti-CD4–Brilliant Violet 510 (clone RM 4.5; BioLegend), anti-CD4–Brilliant Violet 786 (clone GK1.5; BioLegend), 1:100 dilutions of anti-CD4–PerCP (clone GK1.5; BioLegend), or 1:200 dilutions of anti-CD4–FITC (clone RM4.4; BD Biosciences) and 1:600 dilutions of anti-CD44–FITC (clone IM7; eBioscience) and anti-CD8–PerCP-Cy5.5 (clone 53-6.7; eBioscience). Cells were then washed in FACS buffer, permeabilized using the Cytofix/Cytoperm kit (BD Biosciences) according to the manufacturer’s instructions, and stained intracellularly for 30 min at 4°C in dilutions of 1:200 using anti–IFN-γ–PE-Cy7 or anti–IFN-γ–PerCP-Cy5.5 (clone XMG1.2; eBioscience), anti–TNF-α–PE or anti–TNF-α–PeCy7 (clone MP6-XT22; eBioscience), anti–IL-17–allophycocyanin (clone eBio17B7; eBioscience), or dilutions of 1:100 using anti–IL-2–allophycocyanin-Cy7 (clone JES6-5H4; BD Biosciences) mAbs. Cells were subsequently washed with BD Perm/Wash Buffer (BD Biosciences) and resuspended in FACS buffer. Data were collected by running the stained cells on a FACSCanto, FACSCalibur, or FACSFortesa flow cytometer (BD Biosciences) and analyzed using FlowJo software v.10.0.7. In vivo intravascular labeling of T cells At the day of the experiment, mice were injected i.v. with 2 μg of FITC-labeled Abs against CD45.2 in a total volume of 200 μl (clone 102; BioLegend, San Diego, CA). Three minutes after Ab injection, mice were euthanized, and single-cell suspensions were prepared as described above. Adoptive transfer and lung homing For coadoptive transfer studies, donor CD4 T cells from subunit-vaccinated and M. bovis BCG plus subunit–vaccinated animals were isolated by negative selection 3 wk after the last immunization. In brief, cells were isolated from spleen, medial iliac, inguinal, and axillary lymph nodes from eight individual vaccinated donor animals, pooled within the groups and enumerated. Untouched CD4 T cell enrichment was performed from 5 × 108 cells per group using the EasySep Mouse CD4 T cell Enrichment Kit following the manufacturer’s instructions (STEMCELL Technologies). After enrichment, cells were counted, and the density was adjusted to 2.5 × 107 cells per ml for each group (93–95% purity). For tracking, the purified cells were differentially stained for 10 min with 10 μM Cell Proliferation Dye eFluor 450 or 5 μM Cell Proliferation Dye eFluor 670 (Thermo Fisher Scientific). The proliferation dyes were quenched with PBS containing 20% FBS followed by washing and resuspension in PBS. Stained cells were mixed in a ∼1:1 ratio and coadoptively transferred into recipient mice that were infected with M. tuberculosis strain Erdman 3 wk prior. Two hundred microliters (5 × 106 CD4 T cells) was injected into the lateral tail vein of individual recipient mice (the equivalent of one donor mouse per recipient mouse). Eighteen hours after transfer, recipient mice were injected with FITC-labeled anti-CD45.2 Abs for intravascular labeling (clone 102; BD Biosciences) and single-cell suspensions from the lung prepared as described above. Statistical analysis Prism 7 software (GraphPad Prism ver. 8.2.1, San Diego, CA) was used for all statistical analyses. Mean and SEM are indicated for log-transformed CFU counts. Mean and SD are indicated for immune responses. One-way ANOVA combined with Tukey multiple comparison test was used for comparing multiple groups. Statistical significant differences are indicated by asterisks in the figures: *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001. In the guinea pig experiment, the nonparametric log-rank test was used to compare the survival distributions of two samples comparing the survival curves for the vaccinated groups against the saline group. Results ESX-1–associated Ags (M. tuberculosis–specific) provide protection in mice and guinea pigs To investigate the influence of BCG priming on subsequent subunit vaccination, we first designed protective subunit vaccines that selectively incorporated M. tuberculosis–specific Ags, which are not shared with BCG. The M. tuberculosis genome encodes ∼4000 proteins, of which many are potential vaccine targets. However, ∼3900 of these have highly similar orthologs in the BCG genome, which significantly limits the number of potential Ags for this type of vaccine (28, 29). In our selection, we exploited that all BCG substrains lack the genomic locus “region of difference 1” (23), which includes the core genes for the ESX-1 secretion system. In virulent M. tuberculosis strains, ESX-1–secreted proteins are among the most immunogenic Ags and are frequently recognized in TB patients and latently infected individuals (30, 31). However, in BCG, the region of difference 1 deletion is expected to prevent priming of T cells against ESX-1–associated Ags, and we therefore selected among this group of proteins for the first subunit vaccine, referred to as H64. We selected six ESX-1–associated Ags for which proteome studies had identified the proteins in M. tuberculosis culture filtrate or membrane fractions (Table I) and constructed the H64 subunit vaccine as a recombinant fusion protein (Fig. 1A). In H64-vaccinated CB6F1 mice, the CD4 T cells recognized the EsxA, EspD, and EspR Ags (Fig. 1B), and we found that the subunit vaccine induced protection with protein doses ranging from 0.01 to 25 μg per vaccination peaking in the range of 1–5 μg (Fig. 1C). Based on this, an intermediate dose of 2 μg was selected for future mouse studies. Because disease progression and granuloma formation in M. tuberculosis–infected guinea pigs better mimic features of human TB pathology, we also tested the H64 vaccine in Hartley guinea pigs in a long-term infection model. Guinea pigs were challenged with a low dose of virulent M. tuberculosis H37Rv after vaccination with different doses of H64. Animals that reached predefined human end points (weight loss or impact on respiratory rate) were euthanized. Twenty-two weeks after being infected, all animals in the saline control group had been euthanized with a mean survival time of 16.2 wk (SD = ±1.8) (Supplemental Fig. 1A). In comparison, the mean survival time of the M. bovis BCG-vaccinated guinea pigs was 65.1 wk (±8.9). In the four H64-vaccinated groups, the mean survival time ranged from 22.4 wk (±2.3) to 41.6 wk (±9.0) (Fig. 1C). Statistical comparison confirmed that all vaccination groups were better protected than saline-vaccinated animals (p < 0.02, log-rank test). After having confirmed that the ESX-1–associated Ag combination was protective in both mice and guinea pigs, we continued our study of the H64 vaccine by testing its protective efficacy against different challenge strains. Because M. tuberculosis strains harbor genetic diversity that translates into significant differences in Ag diversity, immunogenicity, and virulence, we selected four clinically relevant M. tuberculosis strains belonging to different lineages (2–4) to ensure that the protective signal of H64 was robust and broadly relevant (Fig. 1D, Supplemental Fig. 1B) (32). Similarly to the results with M. tuberculosis Erdman in Fig. 1C, H64 vaccination induced significant protection against all four strains (p < 0.05 or lower) and was equal to or better than the protection obtained with the H56 subunit vaccine that was included to benchmark the new vaccine. In the experiment with H37Rv, H64 was more protective than BCG, but with the other clinical strains, BCG induced similar or better protection than H64 (Fig. 1D). To test how the vaccine performed against a more “aggressive” strain, we challenged mice with M. tuberculosis HN878, which is regarded as hypervirulent because of its rapid growth and induction of severe lung inflammation in mice (33). In this model, both BCG and H64 protected efficiently at week 4 of the infection (p < 0.001), but by week 12, BCG had lost most of its protection, whereas bacterial numbers in H64-vaccinated animals remained significantly lower than BCG as well as the saline control (Fig. 1E, p < 0.005 and p < 0.0001, respectively). View inlineView popup Table I. M. tuberculosis Ags in the H64 fusion protein FIGURE 1. H64 (ESX-1–associated Ags) provide protection against M. tuberculosis in mice and guinea pigs. (A) Illustration of the subunit vaccine H64. The fusion protein is based on ESX-1–associated Ags and does not share Ags with M. bovis BCG. The figure is not drawn to scale. The m.w. of the individual Ags is given in Table I. (B) Ag recognition of splenocytes after immunizing CB6F1 mice with H64 (n = 3). Single-cell cytokine expression was measured by flow cytometry. Any CD4 cell that produced either IFN-γ, TNF-α, and/or IL-2 in response to Ag stimulation was taken as Ag specific. The spleen cells were stimulated with single Ags from H64. EsxH stimulation was included as a negative control (“Control”). Bars and lines illustrate the mean and SD for each Ag. (C) Black curve: protective efficacy studies in CB6F1 mice. Animals were immunized with different doses of H64 in CAF01 adjuvant. The bacterial load was measured in lungs from individual mice 6 wk after M. tuberculosis Erdman challenge (n = 8). The number of bacteria was logarithmic transformed and subtracted from the bacteria numbers in a nonvaccinated control group. Blue curve: protective efficacy studies in guinea pigs (performed at Colorado State University). Guinea pigs were immunized with different doses of H64 in CAF01 and euthanized when predefined humane end points were met after M. tuberculosis infection. Kaplan–Meier survival curves (Supplemental Fig. 1A) were used to estimate the mean survival time for guinea pigs in each of the vaccination groups (n = 10). SDs are shown for each data point. (D) CB6F1 mice immunized with H64, H56, or M. bovis BCG or injected with saline were infected for 6 wk with one of four clinical isolates of M. tuberculosis: H37Rv, Vietnam, Nepal, and Kazakhstan (n = 6–8 per group). MIRU typing in Supplemental Fig. 1B. CFU Log10/lung ± SEM. One-way ANOVA was used for statistical comparison with the saline group for each strain; degree of freedom = 24. (E) H64 or M. bovis BCG immunized or saline-injected C57BL/6 mice were infected 6 wk after immunization with the hypervirulent M. tuberculosis strain Beijing HN878 (performed at Yonsei College of Medicine). After 4- and 12-wk infection, the bacterial burden was measured in the lung (n = 5–8). Mean values and SEMs are illustrated. One-way ANOVA was used for statistical comparison between groups for each time point; degree of freedom = 18. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. In parallel to working with H64, we designed an additional ESX-1 fusion protein in which EspF and PE35 (not immunogenic in H64) were replaced with the Ags EspB and EspA to potentially optimize immunogenicity and/or efficacy (Fig. 2A, Table II). In H74-vaccinated animals, there was a dominant CD4 T cell response to EspB and an increased recognition of EsxA and EspD (Fig. 2B). Similar to H64, H74 induced protective efficacy over a broad range of vaccination doses peaking between 1 and 5 μg (Supplemental Fig. 1C). Because both vaccines were designed to be used in BCG-primed animals, we did a direct head-to-head comparison in this setting. After challenge with M. tuberculosis Erdman, both vaccines induced robust protection on top of BCG at all the measured time points (Fig. 2C). However, at the late time point (20 wk postinfection), lung bacterial numbers were reduced by 1.7 log10 in the BCG control group (p < 0.0001), 1.88 log10 in the BCG-H64–vaccinated group, and 2.22 log10 in the BCG-H74–vaccinated group compared with the saline group. The bacterial burden was thus significantly lower in the H74-vaccinated animals (p < 0.01), and H74 was selected as the ESX-1 vaccine for further studies. FIGURE 2. Improved Ag recognition and protection of H74 compared with H64. (A) Illustration of the subunit vaccine H74. The fusion protein is based on ESX-1–associated Ags. Ags shared with H64 are in light green. The length in amino acids of the individual Ags is given in Table I. (B) Ag recognition of splenocytes isolated from H74-immunized CB6F1 mice (n = 4). The cells were stimulated with single Ags from H74, and EsxH stimulation was included as negative control (Control). CD4 T cells producing either IFN-γ, TNF-α, and/or IL-2 in response to Ag stimulation were taken as Ag specific. Means and SDs are shown for each Ag. (C) Six months after being M. bovis BCG–vaccinated, CB6F1 mice were divided into three groups and vaccinated with either H64 or H74 or saline injected. An age-matched control group was included that did not receive any of the vaccines. All animals were aerosol infected with M. tuberculosis Erdman, and the number of mycobacteria was measured in individual lungs from immunized and nonimmunized mice 6, 12, and 20 wk postinfection (n = 8 per time point). Vertical lines illustrate SEMs. One-way ANOVA was used for statistical comparison between groups at the late time point; degree of freedom = 28. **p < 0.01, ****p < 0.0001. View inlineView popup Table II. M. tuberculosis Ags in the H74 fusion protein In summary, we designed two novel vaccines based exclusively on ESX-1–associated Ags H64 and H74 which demonstrated robust protection in both mice and guinea pigs. H74 was selected for further studies in BCG-primed animals. In BCG-vaccinated mice, ESX-1–associated Ags induce less differentiated CD4 T cells and improve protection compared with BCG boosting It has been demonstrated that BCG vaccination induces highly differentiated T cells (34), but it has not been systematically investigated how this influences T cell quality and protection of subsequent subunit vaccine boosters. We approached this issue by first comparing the protective efficacy of the ESX-1–based H74 vaccine and a BCG booster vaccine (H65), described in a previous study (21). H65 consists of six EsxA family proteins related to ESX-2, -3, or -5 that are all present in BCG (Fig. 3A). Naive mice were immunized with either H65 or H74 as standalone vaccines. Six weeks after M. tuberculosis aerosol challenge, both vaccines reduced the bacterial load by more than 1.0 log10 relative to nonvaccinated animals (p < 0.0001) with no statistical difference between them (Fig. 3B). Having confirmed that the two vaccines induced similar levels of protection in naive mice, we continued by comparing their efficacy in mice that were BCG primed 6 mo prior to subunit vaccination. Twelve weeks after M. tuberculosis Erdman challenge, BCG vaccination reduced the lung bacterial number by 0.85 log10 (p < 0.05). In two separate experiments, H65 boosting did not add significantly to this protection (Fig. 3C, top, Supplemental Fig. 2A), whereas vaccination with the H74 vaccine enhanced the BCG-induced protection (p < 0.05), resulting in a 1.78 log10 reduction of the number of bacteria relative to the nonvaccinated group (p < 0.0001). This observation was robust as similar results were found in a second study with the clinical strain M. tuberculosis Kazakhstan (Fig. 3C, bottom), showing consistently that although the protection was equal in naive mice, the ESX-1 vaccine (M. tuberculosis–specific) induced better protection than the BCG boosting vaccine in BCG-primed animals. FIGURE 3. H74 vaccination (ESX-1–associated Ags) improve protection in BCG-primed animals and induce less differentiated CD4 T cells compared with BCG boosting (H65). (A) Illustration of the BCG booster vaccine H65. All six Ags are shared with M. bovis BCG (21). (B) Groups of naive CB6F1 mice were vaccinated with H74 or H65, and control groups received either a BCG vaccination or saline injections (n = 8). The bacterial numbers were enumerated in lungs 6 wk after an aerosol M. tuberculosis Erdman infection. Means and SEMs are shown by bars and lines. One-way ANOVA was used for statistical analysis; degree of freedom = 27. (C) CB6F1 mice were BCG vaccinated followed by a resting period of 6 mo before being vaccinated with either H74 or H65 (n = 7–8). The bacterial numbers were measured in individual animals 12 wk after they were infected with M. tuberculosis strain Erdman (top) or 25 wk after M. tuberculosis Kazakhstan infection (bottom). One-way ANOVA was used for statistical analysis; degree of freedom = 25 and 26. (D) Timeline for measuring T cell responses in vaccinated CB6F1 mice pre– and post–M. tuberculosis Erdman challenge in the BCG prime–boost model. (E) Splenocytes isolated from CB6F1 mice vaccinated with BCG alone or boosted with H65 were stimulated with the H65 fusion protein. In parallel, splenocytes from BCG-vaccinated animals complemented with H74 were stimulated with the H74 fusion protein. Single-cell expression of cytokine IFN-γ, TNF-α, and IL-2 was measured by flow cytometry, and the frequencies of activated (CD44high) CD4 T cells expressing any of the possible combinations of cytokines are shown in a bar plot for each vaccination group with mean and SD indicated (n = 3). Means (gray bars) and SDs (vertical lines) are shown. The pies are a simplified view of the data illustrating cytokine coexpression patterns of the specific CD4 T cells. The five identified subgroups of cytokine-producing CD4 T cells were as follows: light blue, TNF-α+; dark blue, TNF-α+ and IL-2+; green, TNF-α+, IL-2+, and IFN-γ+; orange, IFN-γ+ and TNF-α+; and red, IFN-γ+. The dotted arches illustrate the fraction of specific CD4 T cells that produced IFN-γ (red) or did not produce cytokine IFN-γ (blue) in response to ex vivo Ag stimulation. The FDS was calculated as the ratio of IFN-γ producers/IFN-γ nonproducers as previously described (6). (F) Cytokine expression profiles were measured in spleens before and in lungs after M. tuberculosis Erdman infection, and the associated FDS score was calculated for each time point and vaccination group (n = 3–4 per time point). Filled circles and vertical bars represents means and SDs. **p < 0.01, ***p < 0.001, ****p < 0.0001. Next, we compared the phenotype of the subunit-specific CD4 T cell response among the vaccinated groups before and after M. tuberculosis challenge (Fig. 3D). We used the individual CD4 T cell cytokine expression profile as a specific and sensitive measure to assess the degree of differentiation (35). For each group, we calculated a simple functional differentiation score (FDS) based on the ratio of IFN-γ producers and nonproducers as has previously been suggested (6). In BCG-vaccinated animals, we found the highest degree of T cell differentiation (FDS = 4.0) with the majority of responding CD4 T cells expressing IFN-γ either alone or in combination with TNF-α and/or IL-2 (Fig. 3E, top). In the H65-boosted group, there was almost a 3-fold increase in the percentage of H65-specific CD4 T cells compared with BCG alone, but H65 boosting induced only minor changes in the cytokine expression profile of the CD4 T cells (FDS = 2.8, Fig. 3E, middle). In contrast, H74 vaccination induced CD4 T cells with a lower degree of differentiation with almost half of the responding T cells expressing TNF-α alone or TNF-α and IL-2 in combination (FDS = 1.0, Fig. 3E, bottom). After M. tuberculosis Erdman infection, the CD4 T cells recruited to the lung maintained an FDS score of ∼ 4.0 in BCG-vaccinated mice during the initial phase of the infection. However, this increased to 10.7 after 6 wk and to 26.1 after 12 wk of infection, clearly showing a further differentiation of the T cell pool during TB infection (Fig. 3F, top). In the H65-boosted group, there was a delay in the differentiation of the CD4 T cells, but at the late time point, the FDS score had increased to 12.8 (Fig. 3F, middle). In contrast, the FDS score for the vaccine-specific CD4 T cells remained around ∼1.0 for all time points in the H74-vaccinated group (Fig. 3F, bottom). Thus, the pool of H74-specific CD4 T cells effectively resisted infection-driven differentiation throughout a 12-wk infection period. We further investigated this in a follow-up study, in which BCG-primed animals were immunized simultaneously with H74 and H65 so that each animal served as its own internal control. In these animals, the H65-specific CD4 T cells had a mean FDS of 3.0 compared with 0.84 for the H74-specific CD4 T cells, confirming that BCG boosting leads to higher T cell differentiation than vaccination with M. tuberculosis–specific Ags (Supplemental Fig. 2B). Finally, ESAT-6 has been shown to be essential for postexposure protection (16), and of relevance to the vaccination of M. tuberculosis–exposed individuals, H74 vaccination induced less differentiated T cells and lower bacterial burdens in the modified Cornell model of latent TB infection, supporting ESX-1–based vaccines for this application (27, 36) (Supplemental Fig. 2C). In summary, in BCG-primed mice, H65 vaccination did neither lead to substantial improvements in T cell differentiation nor did it add significantly to the protection induced by BCG. Conversely, vaccination with ESX-1–associated Ags (H74) induced CD4 T cells with a low differentiation score, which remained low during M. tuberculosis infection. This correlated with a significantly increased protective efficacy. In BCG-vaccinated mice, ESX-1–associated Ags induce CD4 T cells with superior lung-homing capacity Recent studies directly link T cell differentiation status to the ability to enter the infected lung parenchyma and restrict mycobacterial growth (7, 9, 15, 37). In H56/CAF01-vaccinated mice, we have previously shown that lung parenchymal CD4 T cells (protected from anti-CD45 i.v. stain) are less differentiated and express increased levels of the parenchymal homing marker, CXCR3 (15). For this study, to directly link FDS with lung parenchymal trafficking, we first confirmed that there was a strong correlation between FDS and i.v. CD45 labeling in vaccinated animals (Supplemental Fig. 3). Because our data this far showed that pre-existing BCG immunity had a major influence on the differentiation status of subunit-specific CD4 T cells, we next investigated the impact of BCG boosting on lung-homing capacity. For this, we performed an adoptive transfer experiment using donor cells from H74- and H65-vaccinated mice with or without BCG priming. Before cell transfer, we confirmed that all four groups had a solid vaccine-specific CD4 T cell response and used the cytokine expression profiles to determine the degree of CD4 T cell differentiation (Fig. 4A, 4B). Importantly, the T cell differentiation was similar after H74 and H65 vaccination when administered as standalone vaccines, confirming there was no inherent difference in the priming capability of these vaccines. However, in BCG-primed mice, H65 boosting led to a higher T cell differentiation than H74 vaccination, as previously observed (Fig. 4B). FIGURE 4. H74 (ESX-1–associated Ags) induces CD4 T cells with superior lung-homing capacity in BCG-vaccinated mice. (A) Eight weeks after being BCG vaccinated, CB6F1 mice were vaccinated with H65 or H74. Three weeks later, splenocytes were stimulated with H65 or H74 fusion protein, and the expression of cytokines IFN-γ, IL-2, and TNF-α was measured by flow cytometry. CD4 T cells that produced at least one of the cytokines in response to Ag stimulation was regarded as “Ag specific” (n = 4 per group). The mean (gray bar) and SD is shown for each group. (B) The FDS score was calculated as the ratio of IFN-γ+/IFN-γ− cells based on the cytokine expression profile for the individual animal and plotted for each of the four vaccination groups. Horizontal lines represent means, vertical lines, and SDs. One-way ANOVA was used for statistical comparison; degree of freedom = 12. (C) The influence of BCG priming on the lung-homing capacity of H65- and H74-induced CD4 T cells. CD4 T cells were purified from spleens and lymph nodes and pooled within the vaccination groups before labeling with tracker dyes to distinguish cells from donor animals with and without BCG priming. After labeling, cells were mixed 1:1 (e.g., naive plus H65:BCG plus H65) and adoptively transferred to M. tuberculosis Erdman–infected mice. The next day, intravascular localized cells in the recipient mice were labeled using FITC CD45.2, and purified lung cells were stimulated with fusion proteins to identify cytokine expressing (“Ag-specific”) CD4 T cells. For the entire gating strategy, see Supplemental Fig. 4. (D) For each of the four vaccination groups, the percentage of vaccine-specific donor cells entering into the lung parenchyma was calculated, and the influence of BCG priming on vaccine-specific homing was compared for the two subunit vaccines. The lines connect measurements from the same recipient mouse (n = 4). The statistical comparison was done by two-tailed t tests. *p < 0.05. To compare the ability of the vaccine-specific CD4 T cells to enter infected lung parenchyma, we transferred mixed populations of CD4 T cells into M. tuberculosis–infected recipients. Donor CD4 T cells from the four vaccination groups were isolated by negative selection from spleens and inguinal lymph nodes and stained either with Cell Proliferation Dye eFluor 450 or Cell Proliferation Dye eFluor 670 to distinguish cells from BCG-primed animals and cells from animals receiving the subunit vaccine as a standalone. Stained cells were mixed in a ∼1:1 ratio (subunit:BCG prime plus subunit) and cotransferred into M. tuberculosis Erdman–infected recipients in a total 5 × 106 donor CD4 T cells per recipient. Eighteen hours after transfer, recipient mice were injected with FITC-labeled anti-CD45.2 for intravascular labeling, and lung cells were harvested for flow cytometric analysis based on CDP450/670 as well as cytokine staining following H65/H74 stimulation (Fig. 4C, Supplemental Fig. 4). In mice receiving H65-specific cells, only 39.6–72.0% (mean 64%) of the cells from the BCG-H65–boosted donor mice were located in the lung parenchyma, whereas the range was 79.3–99.4% (mean 90%) for donor cells from H65-only vaccinated mice (Fig. 4D). In contrast, we found no significant differences in the percentage of H74-specific CD4 T cells in the parenchyma regardless of whether the cells came from H74 only or BCG-H74–vaccinated animals (range 78.0–99.4 and 72.0–99.4%, respectively, with means of 85.0 and 84.2%). These results clearly demonstrate that pre-existing BCG immunity significantly impacts the functionality of the T cells induced by subunit booster vaccines and that this mechanism can be efficiently bypassed by designing vaccines that selectively incorporate BCG-complementing TB Ags. Discussion The capacity of CD4 T cells to protect against M. tuberculosis is governed by their differentiation state and ability to localize to the site of infection (37). M. bovis BCG vaccination primes a polyfunctional CD4 T cell response that differentiates over time and gradually loses the ability to produce IL-2, proliferate, and to localize to the site of infection, which results in a loss of long-term protective efficacy (11–13). Heterologous prime–boost strategies, in which BCG vaccination is followed by a subunit vaccine boost, are aiming at improving the BCG-induced adaptive immunity in terms of magnitude, durability, and quality of the CD4 T cell response (38). In this study, we tested whether pre-existing BCG immunity impacts the vaccine response of either a “traditional” BCG booster vaccine (H65) or a complementary vaccine based on ESX-1–associated Ags that are specific for M. tuberculosis (H74 and H64, referred to collectively as “ESX-1 vaccines”). In the initial characterization of the ESX-1 vaccines, we demonstrated significant long-term protection in M. tuberculosis H37Rv–challenged guinea pigs as well as M. tuberculosis Erdman–challenged mice. To extend the study beyond the conventional laboratory M. tuberculosis strains, we challenged vaccinated mice with four different strains of M. tuberculosis belonging to three phylogenetically different lineages. The ESX-1 vaccine induced significant protection against all four strains, suggesting that the vaccine will be effective against a broad range of clinical isolates. Two of the selected isolates were part of the W-Beijing family of M. tuberculosis strains, one of them being the hypervirulent M. tuberculosis Beijing HN878. The Beijing strains are particularly relevant to include in this screening as they are highly prevalent, overrepresented among drug-resistant isolates (39), and significantly associated with HIV coinfection in human cases of TB meningitis (40). In a conventional protection readout at 4 wk postinfection, both the ESX-1 vaccine and BCG were highly protective against M. tuberculosis Beijing HN878. This was also true at the later time point for the ESX-1 vaccine, but in line with earlier studies (27, 41), BCG’s protective efficacy waned at the later stage of infection. In the prime–boost model, our data indicates that BCG vaccination has a major influence on the immune responses induced by subsequent subunit vaccines, depending on whether the Ags are shared with BCG or not. Specifically, we observed that boosting BCG with H65 only marginally changed the specific CD4 T cell differentiation status with little or no improvement of the protection. In contrast, BCG priming had minimal influence on the differentiation and functionality of CD4 T cells induced by the ESX-1 vaccine (not sharing Ags with BCG). The ESX-1 vaccine–specific T cells retained their differentiation status after M. tuberculosis infection, and their ability to enter the infected lung parenchyma was increased compared with the T cells in the H65-boosted group. As a result, the ESX-1 vaccine significantly augmented an already strong BCG-induced protection, and we obtained 1.8–2.9 log10 reduction in the lung bacterial load. Importantly, the BCG-priming vaccine was administered more than 6 mo prior to subunit vaccination, suggesting that it was the BCG “T cell imprint,” rather than ongoing bacterial multiplication, that influenced the response of the subunit booster vaccine. In other words, the BCG-specific CD4 T cells appeared to be “locked” with minimal capacity for reprograming into potentially more favorable phenotypes 6 mo later. This is likely because highly differentiated Th1 cells exhibit limited functional plasticity (42, 43) and that subunit boosting merely expands the existing pool (or a subset) of BCG-imprinted CD4 T cells. In addition, BCG may also induce a specific regulatory T cell response that could influence booster vaccines, although this was not investigated in this study (44, 45). Regardless, de novo priming of CD4 T cells using ESX-1–associated (or other M. tuberculosis–specific) Ags bypass this issue, which could be a useful strategy to increase durable protection in BCG-vaccinated populations. A limitation of this study was a lack of specific homing-related makers in the analysis, and in future studies, it will be important to establish whether vaccine-induced reduction of T cell differentiation leads to increased expression of such markers and/or increased contact between Ag-specific CD4 T cells and M. tuberculosis–infected macrophages in the lung. The mouse model has been extensively used to evaluate new prime–boost strategies using M. bovis BCG and subunit vaccines. The results have varied from no additional protection to almost 2 log10 protection compared with the BCG control group (46–48). It is difficult to do a comparative evaluation of these results as the studies differ with regard to vaccine design, mouse strain, M. tuberculosis challenge strain, BCG strain, and dose as well as the interval between prime–boost, boost–challenge, and challenge–sacrifice. However, by evaluating eight different available studies with BCG booster vaccines, we observed that the added protection of BCG boosting only was significant when the BCG-induced protection in itself was low (<0.5 log10) (47, 49–55). In light of our results, one explanation for this could be that a poor BCG vaccine take (or a waning response) will open up for better priming of less differentiated T cell response by the subunit vaccine. In humans, BCG will in most cases be administered to infants, and future booster vaccines are intended to be administered 10–15 y later and preferably before the protective efficacy of BCG wanes. It is not clear how pre-existing BCG immunity will influence subunit vaccination in this setting, and exposure to M. tuberculosis is also likely to play a dominant role in high-endemic areas. In this regard, results demonstrating that subunit vaccines can build on pre-existing M. tuberculosis immunity have been obtained in the recent phase IIb trial, in which M72/AS01e induced 49.7% vaccine efficacy against pulmonary disease in quantiferon positive individuals after 3 y of follow-up (56). Encouragingly, in this study, we also demonstrate that H74 vaccination significantly reduced bacterial burden in the modified Cornell model of postexposure vaccination. Similarly, regarding BCG-vaccinated quantiferon negative individuals, a recent study showed that it is possible to boost BCG protection with a subunit vaccine in adolescents and adults to some extent (57). In this study, H4:IC31 boosting led to 30.1% vaccine efficacy against sustained quantiferon conversion, and based on our data, we speculate that subunit vaccines with ESX-1–associated Ags have the potential to further improve on this result. Additionally, in the study by Nemes et al., BCG revaccination showed a vaccine efficacy of 40.5%, which has sparked renewed interest in using BCG revaccination as a readily applicable intervention (58). In such settings, the influence of recent “BCG imprinting” is likely to be significantly higher if BCG revaccination is to be combined with future subunit vaccines. In conclusion, mycobacterial priming by BCG vaccination induces highly differentiated CD4 T cells that, for at least 6 mo in the mouse model, restrict subsequent booster vaccines in priming additional protective T cells with sufficient memory and lung-homing potential. This phenomenon can efficiently be bypassed by designing vaccines with M. tuberculosis–specific Ags, like the ESX-1–associated Ags studied in this report. We suggest that future studies explore these findings in the human setting, in which it could also be investigated whether exposure to non-tuberculousis mycobacteria play a role in maintaining BCG immunity/T cell “imprint.” Disclosures C.A., N.P.H.K., I.S., E.H.K., T.L., E.M.A., M.R., I.R., P.A., and R.M. are employed by Statens Serum Institut, a nonprofit government research facility of which H56, H64, and H74 and the CAF01 adjuvant are proprietary products. C.A., P.A., and R.M. are coinventors of patents covering ESX-1–based vaccines. Acknowledgments We thank Joshua Woodworth for input on data interpretation and gratefully acknowledge Vivi Andersen and Camilla Rasmussen at Statens Serum Institut for excellent technical assistance. Footnotes This work was supported by The Danish Research Council (DFF - 7016-00310), the National Institutes of Health/National Institute of Allergy and Infectious Diseases (Grant 1R01AI135721-01), the European Union’s Horizon 2020 Framework Programme for Research and Innovation under Grant Agreement 643381 as part of the TBVAC2020 Consortium, and the National Institutes of Health/National Institute of Allergy and Infectious Diseases program Advanced Small Animal Models for the Testing of Candidate Therapeutic and Preventative Interventions against Mycobacteria (HHSN272201000009I-003, Task Order 12) at Colorado State University. The online version of this article contains supplemental material. Abbreviations used in this article: BCG Bacillus Calmette–Guérin FDS functional differentiation score MIRU mycobacterial interspersed repetitive unit TB tuberculosis. Received May 18, 2020. Accepted August 5, 2020. Copyright © 2020 by The American Association of Immunologists, Inc. This article is distributed under The American Association of Immunologists, Inc., Reuse Terms and Conditions for Author Choice articles. References ↵WHO. 2018. Global Tuberculosis Report 2018. World Health Organization, Geneva, Switzerland. ↵Trunz, B. B., P. Fine, C. Dye. 2006. Effect of BCG vaccination on childhood tuberculous meningitis and miliary tuberculosis worldwide: a meta-analysis and assessment of cost-effectiveness. Lancet 367: 1173–1180.OpenUrlCrossRefPubMed ↵Mangtani, P., I. Abubakar, C. Ariti, R. Beynon, L. Pimpin, P. E. M. Fine, L. C. Rodrigues, P. G. Smith, M. Lipman, P. F. Whiting, J. A. Sterne. 2014. Protection by BCG vaccine against tuberculosis: a systematic review of randomized controlled trials. Clin. Infect. Dis. 58: 470–480.OpenUrlCrossRefPubMed ↵Leveton, C., S. Barnass, B. Champion, S. Lucas, B. De Souza, M. Nicol, D. Banerjee, G. Rook. 1989. T-cell-mediated protection of mice against virulent Mycobacterium tuberculosis. Infect. Immun. 57: 390–395. ↵Green, A. M., R. Difazio, J. L. Flynn. 2013. IFN-γ from CD4 T cells is essential for host survival and enhances CD8 T cell function during Mycobacterium tuberculosis infection. J. Immunol. 190: 270–277. ↵Moguche, A. O., M. Musvosvi, A. Penn-Nicholson, C. R. Plumlee, H. Mearns, H. Geldenhuys, E. Smit, D. Abrahams, V. Rozot, O. Dintwe, et al. 2017. Antigen availability shapes T cell differentiation and function during tuberculosis. Cell Host Microbe 21: 695–706.e5.OpenUrlCrossRefPubMed ↵Sallin, M. A., S. Sakai, K. D. Kauffman, H. A. Young, J. Zhu, D. L. Barber. 2017. Th1 differentiation drives the accumulation of intravascular, non-protective CD4 T cells during tuberculosis. Cell Rep. 18: 3091–3104.OpenUrlCrossRef ↵He, H., P. N. Nehete, B. Nehete, E. Wieder, G. Yang, S. Buchl, K. J. Sastry. 2011. Functional impairment of central memory CD4 T cells is a potential early prognostic marker for changing viral load in SHIV-infected rhesus macaques. PLoS One 6: e19607. ↵Sakai, S., K. D. Kauffman, J. M. Schenkel, C. C. McBerry, K. D. Mayer-Barber, D. Masopust, D. L. Barber. 2014. Cutting edge: control of Mycobacterium tuberculosis infection by a subset of lung parenchyma-homing CD4 T cells. J. Immunol. 192: 2965–2969. ↵Srivastava, S., J. D. Ernst. 2013. Cutting edge: direct recognition of infected cells by CD4 T cells is required for control of intracellular Mycobacterium tuberculosis in vivo. J. Immunol. 191: 1016–1020. ↵Lindenstrøm, T., A. Moguche, M. Damborg, E. M. Agger, K. Urdahl, P. Andersen. 2018. T cells primed by live mycobacteria versus a tuberculosis subunit vaccine exhibit distinct functional properties. EBioMedicine 27: 27–39.OpenUrl Perdomo, C., U. Zedler, A. A. Kühl, L. Lozza, P. Saikali, L. E. Sander, A. Vogelzang, S. H. E. Kaufmann, A. Kupz. 2016. Mucosal BCG vaccination induces protective lung-resident memory T cell populations against tuberculosis. mBio 7: e01686-16. ↵Orme, I. M. 2010. The Achilles heel of BCG. Tuberculosis (Edinb.) 90: 329–332.OpenUrlCrossRef ↵Rodo, M. J., V. Rozot, E. Nemes, O. Dintwe, M. Hatherill, F. Little, T. J. Scriba. 2019. A comparison of antigen-specific T cell responses induced by six novel tuberculosis vaccine candidates. PLoS Pathog. 15: e1007643. ↵Woodworth, J. S., S. B. Cohen, A. O. Moguche, C. R. Plumlee, E. M. Agger, K. B. Urdahl, P. Andersen. 2017. Subunit vaccine H56/CAF01 induces a population of circulating CD4 T cells that traffic into the Mycobacterium tuberculosis-infected lung. Mucosal Immunol. 10: 555–564.OpenUrlCrossRefPubMed ↵Hoang, T., C. Aagaard, J. Dietrich, J. P. Cassidy, G. Dolganov, G. K. Schoolnik, C. V. Lundberg, E. M. Agger, P. Andersen. 2013. ESAT-6 (EsxA) and TB10.4 (EsxH) based vaccines for pre- and post-exposure tuberculosis vaccination. PLoS One 8: e80579. Billeskov, R., T. Lindenstrøm, J. Woodworth, C. Vilaplana, P. J. Cardona, J. P. Cassidy, R. Mortensen, E. M. Agger, P. Andersen. 2018. High antigen dose is detrimental to post-exposure vaccine protection against tuberculosis. Front. Immunol. 8: 1973.OpenUrl Henao-Tamayo, M., G. S. Palaniswamy, E. E. Smith, C. A. Shanley, B. Wang, I. M. Orme, R. J. Basaraba, N. M. DuTeau, D. Ordway. 2009. Post-exposure vaccination against Mycobacterium tuberculosis. Tuberculosis (Edinb.) 89: 142–148.OpenUrl Turner, J., E. R. Rhoades, M. Keen, J. T. Belisle, A. A. Frank, I. M. Orme. 2000. Effective preexposure tuberculosis vaccines fail to protect when they are given in an immunotherapeutic mode. Infect. Immun. 68: 1706–1709. ↵Taylor, J. L., O. C. Turner, R. J. Basaraba, J. T. Belisle, K. Huygen, I. M. Orme. 2003. Pulmonary necrosis resulting from DNA vaccination against tuberculosis. Infect. Immun. 71: 2192–2198. ↵Knudsen, N. P., S. Nørskov-Lauritsen, G. M. Dolganov, G. K. Schoolnik, T. Lindenstrøm, P. Andersen, E. M. Agger, C. Aagaard. 2014. Tuberculosis vaccine with high predicted population coverage and compatibility with modern diagnostics. Proc. Natl. Acad. Sci. USA 111: 1096–1101. ↵Aguilo, N., J. Gonzalo-Asensio, S. Alvarez-Arguedas, D. Marinova, A. B. Gomez, S. Uranga, R. Spallek, M. Singh, R. Audran, F. Spertini, C. Martin. 2017. Reactogenicity to major tuberculosis antigens absent in BCG is linked to improved protection against Mycobacterium tuberculosis. Nat. Commun. 8: 16085.OpenUrlCrossRef ↵Pym, A. S., P. Brodin, L. Majlessi, R. Brosch, C. Demangel, A. Williams, K. E. Griffiths, G. Marchal, C. Leclerc, S. T. Cole. 2003. Recombinant BCG exporting ESAT-6 confers enhanced protection against tuberculosis. Nat. Med. 9: 533–539.OpenUrlCrossRefPubMed Gröschel, M. I., F. Sayes, S. J. Shin, W. Frigui, A. Pawlik, M. Orgeur, R. Canetti, N. Honoré, R. Simeone, T. S. van der Werf, et al. 2017. Recombinant BCG expressing ESX-1 of Mycobacterium marinum combines low virulence with cytosolic immune signaling and improved TB protection. Cell Rep. 18: 2752–2765.OpenUrlCrossRef ↵Bottai, D., W. Frigui, S. Clark, E. Rayner, A. Zelmer, N. Andreu, M. I. de Jonge, G. J. Bancroft, A. Williams, P. Brodin, R. Brosch. 2015. Increased protective efficacy of recombinant BCG strains expressing virulence-neutral proteins of the ESX-1 secretion system. Vaccine 33: 2710–2718.OpenUrlCrossRefPubMed ↵Aagaard, C. S., T. T. Hoang, C. Vingsbo-Lundberg, J. Dietrich, P. Andersen. 2009. Quality and vaccine efficacy of CD4+ T cell responses directed to dominant and subdominant epitopes in ESAT-6 from Mycobacterium tuberculosis. J. Immunol. 183: 2659–2668. ↵Aagaard, C., T. Hoang, J. Dietrich, P. J. Cardona, A. Izzo, G. Dolganov, G. K. Schoolnik, J. P. Cassidy, R. Billeskov, P. Andersen. 2011. A multistage tuberculosis vaccine that confers efficient protection before and after exposure. Nat. Med. 17: 189–194.OpenUrlCrossRefPubMed ↵Cole, S. T., R. Brosch, J. Parkhill, T. Garnier, C. Churcher, D. Harris, S. V. Gordon, K. Eiglmeier, S. Gas, C. E. Barry III., et al. 1998. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393: 537–544.OpenUrlCrossRefPubMed ↵Behr, M. A., M. A. Wilson, W. P. Gill, H. Salamon, G. K. Schoolnik, S. Rane, P. M. Small. 1999. Comparative genomics of BCG vaccines by whole-genome DNA microarray. Science 284: 1520–1523. ↵Millington, K. A., S. M. Fortune, J. Low, A. Garces, S. M. Hingley-Wilson, M. Wickremasinghe, O. M. Kon, A. Lalvani. 2011. Rv3615c is a highly immunodominant RD1 (region of difference 1)-dependent secreted antigen specific for Mycobacterium tuberculosis infection. Proc. Natl. Acad. Sci. USA 108: 5730–5735. ↵van Pinxteren, L. A., P. Ravn, E. M. Agger, J. Pollock, P. Andersen. 2000. Diagnosis of tuberculosis based on the two specific antigens ESAT-6 and CFP10. Clin. Diagn. Lab. Immunol. 7: 155–160.OpenUrlCrossRefPubMed ↵Coscolla, M., S. Gagneux. 2010. Does M. tuberculosis genomic diversity explain disease diversity? Drug Discov. Today Dis. Mech. 7: e43–e59.OpenUrlCrossRefPubMed ↵Manca, C., L. Tsenova, S. Freeman, A. K. Barczak, M. Tovey, P. J. Murray, C. Barry, G. Kaplan. 2005. Hypervirulent M. tuberculosis W/Beijing strains upregulate type I IFNs and increase expression of negative regulators of the Jak-Stat pathway. J. Interferon Cytokine Res. 25: 694–701.OpenUrlCrossRefPubMed ↵Nandakumar, S., S. Kannanganat, J. E. Posey, R. R. Amara, S. B. Sable. 2014. Attrition of T-cell functions and simultaneous upregulation of inhibitory markers correspond with the waning of BCG-induced protection against tuberculosis in mice. PLoS One 9: e113951. ↵Seder, R. A., P. A. Darrah, M. Roederer. 2008. T-cell quality in memory and protection: implications for vaccine design. [Published erratum appears in 2008 Nat. Rev. Immunol. 8: 486.] Nat. Rev. Immunol. 8: 247–258.OpenUrlCrossRefPubMed ↵McCune, R. M. Jr.., W. McDermott, R. Tompsett. 1956. The fate of Mycobacterium tuberculosis in mouse tissues as determined by the microbial enumeration technique. II. The conversion of tuberculous infection to the latent state by the administration of pyrazinamide and a companion drug. J. Exp. Med. 104: 763–802.OpenUrlAbstract ↵Sakai, S., K. D. Mayer-Barber, D. L. Barber. 2014. Defining features of protective CD4 T cell responses to Mycobacterium tuberculosis. Curr. Opin. Immunol. 29: 137–142.OpenUrlCrossRefPubMed ↵Lewinsohn, D. A., D. M. Lewinsohn, T. J. Scriba. 2017. Polyfunctional CD4+ T cells as targets for tuberculosis vaccination. Front. Immunol. 8: 1262.OpenUrlCrossRefPubMed ↵Glynn, J. R., J. Whiteley, P. J. Bifani, K. Kremer, D. van Soolingen. 2002. Worldwide occurrence of Beijing/W strains of Mycobacterium tuberculosis: a systematic review. Emerg. Infect. Dis. 8: 843–849.OpenUrlCrossRefPubMed ↵Caws, M., G. Thwaites, K. Stepniewska, T. N. Nguyen, T. H. Nguyen, T. P. Nguyen, N. T. Mai, M. D. Phan, H. L. Tran, T. H. Tran, et al. 2006. Beijing genotype of Mycobacterium tuberculosis is significantly associated with human immunodeficiency virus infection and multidrug resistance in cases of tube
|
|
Scooped by
Gilbert C FAURE
September 21, 2020 9:18 AM
|
Abstract Understanding the mechanisms involved in induction and regulation of the immune and inflammatory response to Helicobacter pylori is extremely important in determining disease outcomes. H p...
|
|
|
Scooped by
Gilbert C FAURE
January 19, 2022 4:59 AM
|
BackgroundData regarding symptoms in the lactating mother-infant dyad and their immune response to COVID-19 mRNA vaccination during lactation are needed to inform vaccination guidelines.MethodsFrom a prospective cohort of 50 lactating individuals who received mRNA-based vaccines for COVID-19...
|
|
Scooped by
Gilbert C FAURE
December 30, 2021 4:16 AM
|
COVID-19 mRNA vaccines are highly effective at preventing COVID-19. Prior studies have found detectable SARS-CoV-2 IgG antibodies in oral mucosal specimens of participants with history of COVID-19. To assess the development of oral SARS-CoV-2 IgG antibodies among people who received either the...
|
|
Scooped by
Gilbert C FAURE
September 24, 2021 2:23 PM
|
The coronavirus disease 2019 (COVID-19) pandemic has highlighted the urgent need for effective prophylactic vaccination to prevent the spread of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2).
|
|
Scooped by
Gilbert C FAURE
September 14, 2021 5:14 AM
|
Despite remarkable progress in the development and authorization of vaccines against severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the…...
|
|
Scooped by
Gilbert C FAURE
August 2, 2021 3:58 AM
|
Mucosal vaccines offer the potential to trigger robust protective immune responses at the predominant sites of pathogen infection. In principle, the induction of adaptive immunity at mucosal sites, involving secretory antibody responses and tissue-resident T cells, has the capacity to prevent an...
|
|
Scooped by
Gilbert C FAURE
May 19, 2021 1:52 PM
|
Fentanyl is a major contributor to the devastating increase in overdose deaths from substance use disorders (SUD). A vaccine targeting fentanyl could be a powerful immunotherapeutic. Here, we evaluated adjuvant and delivery strategies for conjugate antigen vaccination with fentanyl-based haptens.
|
|
Scooped by
Gilbert C FAURE
March 27, 2021 4:17 AM
|
Date 123: 25 March | 3:30PM GMT / 11:30AM EST / 4:30PM CETToday, there are three marketed nasal vaccines available for human use but many more are in development for both human and veterinary use.Nasal vaccination provides an alternative to the more...
|
|
Scooped by
Gilbert C FAURE
February 21, 2021 4:16 AM
|
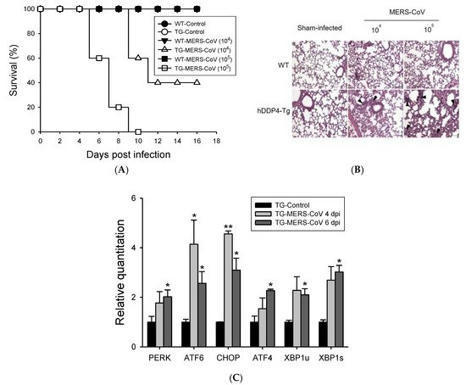
Middle East respiratory syndrome coronavirus (MERS-CoV) causes severe acute respiratory symptoms. Due to the lack of medical countermeasures, effective and safe vaccines against MERS-CoV infection are urgently required.
|
|
Scooped by
Gilbert C FAURE
January 20, 2021 4:37 AM
|
Making “Edible Vaccines” in Plants Some scientists believe that vaccines could be produced in edible plants, such as bananas or potatoes, which would then provide the delivery vehicle. This approach would yield vaccines that are needle-free and require no adjuvants, chemicals that stimulate the immune response. When the plant is ingested, the plant cell walls would protect the vaccine antigens from degradation by stomach acid and digestive enzymes. Then in the intestine, the antigens would be released and transported into the circulatory system. Edible vaccines would stimulate both mucosal and systemic immunity, providing a higher level of protection than traditional vaccines. Advocates claim that vaccines produced in edible plants would be low-cost and would not require refrigeration, making them more accessible to poor people in developing countries. But although several edible vaccines have entered clinical trials, none has yet been approved for marketing.5 Indeed, many technical questions remain to be answered before edible vaccines will become a viable option. Although a potato may express vaccine antigens, how many people will eat a raw potato? If the potato is cooked, will the vaccine remain effective? How does one ensure a uniform product or determine the appropriate dose? Some scientists are now thinking of producing edible vaccines in bananas or potatoes and processing them into a powder, which would be more usable and consistent. It is not yet clear, however, if this approach is commercially viable. In any event, regulatory safeguards will be required to protect consumer health and the environment.
|
|
Scooped by
Gilbert C FAURE
December 9, 2020 9:09 AM
|
Citation: Langel SN, Otero CE, Martinez DR, Permar SR (2020) Maternal gatekeepers: How maternal antibody Fc characteristics influence passive transfer and infant protection. PLoS Pathog 16(3): e1008303. https://doi.org/10.1371/journal.ppat.1008303 Editor: Matthew J. Evans, Mount Sinai School of Medicine, UNITED STATES Published: March 26, 2020 Copyright: © 2020 Langel et al. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Funding: SNL is supported by an NIH National Institute of Allergy and Infectious Diseases (NIAID: https://www.niaid.nih.gov) Ruth L. Kirschstein National Research Service Award T32 AI007392. CEO is supported by an NIH NIAID Ruth L. Kirschstein National Research Service Award T32 CA009111. DRM is supported by an NIH NIAID Ruth L. Kirschstein National Research Service Award T32 AI007151 and a Burroughs Wellcome Postdoctoral Enrichment Award (www.bwfund.org). SRP is supported by NIH NIAID R01 AI122909, P01 AI129859, and P01 AI117915. The funders had no role in study design, data collection, analysis, decision to publish, or preparation of the manuscript. The content is solely the view of the authors and does not necessarily represent the official views of the National Institutes of Health. Competing interests: I have read the journal's policy and have the following conflicts: SRP serves as a consultant for Pfizer, Sanofi, Moderna, and Merck vaccines and has a sponsored program on preclinical cytomegalovirus vaccine development with Merck and Moderna. All other authors declare no competing interests. Introduction Maternal antibodies (MatAbs) passively transferred across the placenta and into breast milk are critical for protection against infectious disease and immune development during the first year of life [1]. Passive transfer in the placenta and mammary gland (MG) is dependent on MatAbs binding to crystallizable fragment (Fc) receptors (FcRs) on polarized epithelial cells. For example, immunoglobulin G (IgG) transfers through the placenta by Fc domain binding to the Fc receptor neonatal (FcRn) on syncytiotrophoblasts [2], providing the fetus with a systemic source of protective IgG antibodies [3]. Additionally, maternal dimeric immunoglobulin A (dIgA) antibodies transfer into breast milk by binding to the polymeric immunoglobulin receptor (pIgR) on MG epithelial cells through the antibody joining chain (J-chain) [4] and provide immune protection in the gut while shaping microbiota colonization [4,5]. Yet MatAbs can interfere with the neonatal immune response, particularly after vaccination [6]. This Pearl explores the role of monomeric IgG, the only antibody isotype to cross the placenta, and polymeric IgA, the major antibody species in breast milk, and their Fc domain characteristics on passive transfer to and functional activity in the newborn. The IgG Fc domain and its effector functions in the context of MatAb passive transfer Antibodies contain 2 domains that exert a wide range of effector functions. The antigen-binding fragment (Fab) domain binds foreign antigens and drives antibody diversity [7], whereas the Fc is responsible for initiating innate immune cell activation and passive antibody transfer [8]. The classical FcRn-driven IgG transport mechanism is responsible for shuttling IgG within acidified endosomes across the syncytiotrophoblast cell barrier from maternal to fetal circulation (Fig 1A) [2]. Once in the neonate, the IgG Fc domain can engage classical type I Fc gamma (Fcγ) receptors (activating [FcγRI, FcγRIIa, FcγRIIc, FcγRIIIa, FcγRIIIb]; inhibitory [FcγRIIb]) or complement to mediate nonneutralizing functions like antibody-dependent cell-mediated cytotoxicity (ADCC) and antibody-dependent cellular phagocytosis (ADCP), or complement-dependent cytotoxicity (CDC), respectively (Fig 1A) [9]. Nonclassical type II FcRs are C-type lectin receptors, including CD209 (DC-SIGN) and CD23, which bind IgG to facilitate immune complex formation [9]. Considering each family of Fc receptors initiates distinct effector functions, the diversity of the Fc domain allows tailoring of nonneutralizing Fc-mediated activity to protect against viruses like HIV, influenza, and cytomegalovirus [10–12]. Alternatively, pathogens such as dengue virus utilize complement and FcR pathways for antibody-dependent enhancement of disease [13]. Download: PPT PowerPoint slide PNG larger image TIFF original image Fig 1. Maternal antibody passive transfer and functional activity in the neonate. (A) IgG passive transfer in the placenta influences FcγR-mediated cell cytotoxicity, phagocytosis, and complement activation in the developing fetus/newborn. (B) IgA passive transfer in the mammary gland results in FcαR- and IgA-mediated cell activation and microbiota regulation, respectively. Fab, antigen-binding fragment; Fc, crystallizable fragment; FcαR, Fc alpha receptor; FcRn, Fc receptor neonatal; FcγR, Fc gamma receptor; IgA, immunoglobulin A; IgG, immunoglobulin G; J-chain, joining chain; pIgR, polymeric immunoglobulin receptor. https://doi.org/10.1371/journal.ppat.1008303.g001 The IgG Fc domain mediates considerable heterogeneity of its effector functions depending on the subclass and glycan profile. For example, each IgG subclass (IgG1-4) has one N-glycosylation site in each CH2 domain, an important binding site for FcγRs (Fig 2). Interestingly, there are up to 36 possible antibody glycan profiles that could theoretically be present on each CH2 domain. This allows for combinatorial diversity of the Fc region with 144 different potential functional states for the 4 IgG subclasses [14]. This is relevant in the context of maternal–fetal immunity, as FcRn has different binding affinities to each IgG subclass, which may reflect their placental transfer efficiency [15]. Additionally, recent data suggest that Fc glycan profiles create antibody transfer hierarchies in the placenta of both healthy and HIV-infected pregnant women. For example, in healthy pregnant women, there is a shift toward IgG galactosylated antibodies, which have higher FcRn-binding affinity, are more efficiently transferred across the placenta, and enhance natural killer (NK) cell degranulation and chemokine secretion [16]. Additionally, binding of tetanus toxoid–specific IgG to placental FcγRIIa H131, FcγRIIa R131, and FcγRIIIa F158 (but not canonical FcRn) was positively associated with placental IgG transfer efficiency in HIV-infected women, suggesting that noncanonical placental FcRs may also play a role in IgG placental transfer [17,18]. Fc-mediated differential selection of IgG antibodies in the placenta is likely an adaptive evolutionary mechanism to passively transfer the most effective antibodies to the infant, which can be altered by disease status. Download: PPT PowerPoint slide PNG larger image TIFF original image Fig 2. Schematic representation of IgA and IgG glycosylation. N-linked glycosylation is depicted as yellow circles, whereas O-linked glycosylation is depicted as green stars. IgA, immunoglobulin A; IgG, immunoglobulin G; sIgA2, secretory IgA. https://doi.org/10.1371/journal.ppat.1008303.g002 Do IgA Fc region characteristics influence IgA passive transfer or effector function in breast milk? IgA antibodies bind their own unique Fc receptors that facilitate epithelial cell transcytosis and innate immune cell activation. dIgA antibodies are composed of 2 monomers, linked by a 15-kDa J-chain. Transport of dIgA into breast milk is dependent on C-terminal binding of the J-chain to a portion of pIgR, known as the secretory component, on the basolateral surface of MG epithelial cells [19]. Without the J-chain, IgA antibodies are secreted as monomers and are not actively transported across the mucosal epithelium [20]. After transport of the J-chain/pIgR complex to the apical portion of the cell, pIgR is cleaved, releasing secretory IgA (sIgA) into breast milk and other mucosal fluids (Fig 1B) [21]. IgA also binds to Fc alpha receptor (FcαR) [22] on the surface of myeloid cells. Monomeric serum IgA induces inhibitory signals, whereas IgA immune complexes have increased avidity to and cross-link FcαRI, resulting in proinflammatory responses [23]. The dominant immunoglobulin class in breast milk sIgA has decreased affinity for FcαRI likely due to steric hindrance from the attached secretory component [24]. Although the opsonic activity of sIgA is poor compared with monomeric and dIgA [25], sIgA can initiate macrophage phagocytosis and neutrophil respiratory burst [26,27]. Further defining the anti- and proinflammatory effects of IgA subclass–FcR interactions would allow fine tuning of breast milk immunity and may represent an attractive therapeutic strategy. Considering breast milk sIgA protects from pathogenic insult and facilitates maturation of the microbiota in early life [28], understanding breast milk antibody Fc–mediated effector functions is integral to neonatal intestinal health (Fig 1B). This is further supported by the fact that bacteria have evolved mechanisms to block IgA and FcαR interactions [29]. Additionally, it is likely that the more complex and extensive glycosylation pattern of IgA antibodies (Fig 2) impacts effector function in milk. Indeed, mucosal secretions, including breast milk, contain mostly IgA2 [30], which has 2 [IgA2m(1)] or 3 [IgA2m(2)] additional conserved N-glycans compared with IgA1 [31], which dominates in serum [32]. Recent evidence demonstrates that IgA glycan–bacteria interactions regulate gut microbiota composition and metabolism as well as retrograde transport of sIgA immune complexes back to the lamina propria independent of antibody–epitope interactions [33–35]. Additionally, the sialic acid in IgA antibody’s C-terminal tail competes with receptor binding to some viruses, providing an innate line of defense against infection [36]. This is relevant to breast milk IgA passive transfer, considering that the leading causes of severe pediatric gastroenteritis worldwide (rotavirus and norovirus) both utilize sialic acid receptors for intestinal infection [37,38]. Studies are needed to define the mechanisms of Fc-mediated IgA effector functions in breast milk, including their interactions with the developing infant microbiome and protection against intestinal viral infections. MatAb interference is influenced by MatAb Fc domain–receptor interactions Despite the well-documented benefits of MatAbs on early life immunity [3], a mounting body of evidence indicates that MatAbs can inhibit immune responses to certain infant vaccinations [6,39]. A recent meta-analysis demonstrated that MatAbs acquired transplacentally inhibited antibody responses to priming vaccinations and these effects were not overcome by administration of a booster dose [40]. This highlights the “window of susceptibility” that exists for infants when MatAbs are not high enough for seroprotection yet still interfere with infant vaccine responses. Multiple mechanisms have been proposed to describe both Fab- and Fc-mediated MatAb interference. These include live virus vaccine neutralization, inhibition of B-cell responses by epitope masking [39], and IgG Fc binding to FcγRIIB [41]. Kim and colleagues demonstrated that B-cell responses to a live attenuated measles vaccine were inhibited by passively transferred measles-specific IgG antibodies in a FcγRIIB-dependent manner, suggesting that IgG Fc region characteristics contribute to suppression of the immune response [41]. Additionally, removing the glycans from IgG2b abolished its immunosuppressive activity both in vitro and in vivo [42,43]. Considering that the mechanisms of MatAb interference likely differ depending on vaccine type (live attenuated, inactivated, subunit), route of delivery (oral, subcutaneous [SQ], intramuscular [IM]), and adjuvant formulation, defining glycan-dependent passive transfer of maternal IgG antibody subclasses in the placenta is crucial for developing effective maternal immunization strategies. Although less studied, IgA antibodies in breast milk may also contribute to interference of immune responses to oral infant vaccines such as rotavirus and poliovirus [44,45]. Indeed, the 2 oral rotavirus vaccines Rotarix and Rotateq demonstrate lower efficacy and immunogenicity in infants from some low- and middle-income countries (LMICs) [46,47] where women tend to have higher titers of antirotavirus IgA antibodies and neutralizing activity in milk [48,49]. The high rotavirus neutralizing activity in breast milk of women from LMICs may partially explain the decrease in rotavirus vaccine efficacy; however, IgA MatAb interference is not well defined [49]. Although infant CD4+ T-cell responses are mostly unaffected by MatAb interference [6,39], recent work demonstrated that MatAbs dampen mucosal T-cell responses against commensal bacteria [50] and limit the expansion of T follicular helper (TFH) cells in the germinal center (GC) [39]. The premature decline in GC TFH cells resulted in the reduction or prevention of plasma cell and memory B-cell generation in a MatAb- and antigen dose–dependent manner [39]. Interestingly, at low or intermediate titers, MatAbs did not prevent the induction of memory B cells, suggesting a gradient effect of MatAbs on infant immune responses [39]. Defining the functional consequences of MatAb gradients will be essential for infant vaccine design and immunization timing. For example, and in addition to previously discussed mechanisms, there is evidence that preexisting antibodies can promote higher affinity antibody responses due to competitive binding in the GC [51] and increased uptake and antigen presentation through immune complexes (ICs) [52] in a Fc glycosylation–dependent manner [53,54]. However, more research is needed to determine the effects of these mechanisms in the setting of passively transferred MatAbs and infant GC responses. Harnessing MatAb Fc region characteristics and receptor interactions to fine-tune maternal immunizations that maximize infant protection Passive transfer of MatAbs is central to pathogen protection and immune system development in early life. However, MatAb-mediated interference dampens antibody responses to vaccinations, leaving children more susceptible to infections while increasing transmission rates to unvaccinated cohorts. Recent work demonstrated that maternal IgG antibodies are differentially transferred across the placenta in an Fc glycan–dependent manner [16,17]. Considering vaccination strategies could direct antigen-specific antibody glycosylation [55], defining MatAb glycan profiles represents an adaptable and powerful mechanism to fine-tune maternal immunizations that maximize infant protection while limiting MatAb interference. Future studies are needed to determine (1) how maternal vaccination and their distinct adjuvant mixtures alter IgG Fc domain glycosylation and whether certain glycan profiles are associated with IgG Fc-mediated MatAb interference and (2) whether or not the IgA Fc domain regulates passive transfer in the MG or effector function in breast milk. Defining the molecular mechanisms of Fc-mediated functional activity at the maternal–fetal/neonatal interface is critical for developing next-generation maternal vaccines and antibody-based therapeutics to improve the health of the mother–neonatal dyad. References 1. Hurley WL, Theil PK. Perspectives on immunoglobulins in colostrum and milk. Nutrients. 2011 Apr;3(4): 442–474. pmid:22254105; PubMed Central PMCID: PMC3257684. View Article PubMed/NCBI Google Scholar 2. Simister NE, Story CM, Chen HL, Hunt JS. An IgG-transporting Fc receptor expressed in the syncytiotrophoblast of human placenta. Eur J Immunol. 1996 July;26(7): 1527–1531. pmid:8766556 View Article PubMed/NCBI Google Scholar 3. Fouda GG, Martinez DR, Swamy GK, Permar SR. The Impact of IgG transplacental transfer on early life immunity. Immunohorizons. 2018 Jan 1;2(1): 14–25. pmid:29457151 View Article PubMed/NCBI Google Scholar 4. Rogier EW, Frantz AL, Bruno MEC, Wedlund L, Cohen DA, Stromberg AJ, et al. Secretory antibodies in breast milk promote long-term intestinal homeostasis by regulating the gut microbiota and host gene expression. Proc Natl Acad Sci U S A. 2014 Feb 25;111(8):3074–3079. pmid:24569806 View Article PubMed/NCBI Google Scholar 5. Sadeharju K, Knip M, Virtanen SM, Savilahti E, Tauriainen S, Koskela P, et al. Maternal antibodies in breast milk protect the child from enterovirus infections. Pediatrics. 2007 May;119: 941–946. pmid:17473095. View Article PubMed/NCBI Google Scholar 6. Niewiesk S. Maternal antibodies: clinical significance, mechanism of interference with immune responses, and possible vaccination strategies. Front Immunol. 2014 Sep 16;5: 446. pmid:25278941; PubMed Central PMCID: PMC4165321. View Article PubMed/NCBI Google Scholar 7. Li Z, Woo CJ, Iglesias-Ussel MD, Ronai D, Scharff MD. The generation of antibody diversity through somatic hypermutation and class switch recombination. Genes Dev. 2014 Jan 1;18(1): 1–11. pmid:14724175. View Article PubMed/NCBI Google Scholar 8. Lu LL, Suscovich TJ, Fortune SM, Alter G. Beyond binding: antibody effector functions in infectious diseases. Nat Rev Immunol. 2018 Jan;18(1):46–61. pmid:29063907 View Article PubMed/NCBI Google Scholar 9. Pincetic A, Bournazos S, DiLillo DJ, Maamary J, Wang TT, Dahan R, et al. Type I and type II Fc receptors regulate innate and adaptive immunity. Nat Immunol. 2014 Aug;15(8):707–16. pmid:25045879 View Article PubMed/NCBI Google Scholar 10. Horwitz JA, Bar-On Y, Lu CL, Fera D, Lockhart AAK, Lorenzi JCC, et al. Non-neutralizing Antibodies Alter the Course of HIV-1 Infection In Vivo. Cell. 2017 Aug 10;170(4):637–648.e10. pmid:28757252 View Article PubMed/NCBI Google Scholar 11. Henry Dunand CJ, Leon PE, Huang M, Choi A, Chromikova V, Ho IY, et al. Both Neutralizing and Non-Neutralizing Human H7N9 Influenza Vaccine-Induced Monoclonal Antibodies Confer Protection. Cell Host Microbe. 2016 Jun 8;19(6):800–813. pmid:27281570 View Article PubMed/NCBI Google Scholar 12. Nelson CS, Huffman T, Jenks JA, Cisneros de la Rosa E, Xie G, Vandergrift N, et al. HCMV glycoprotein B subunit vaccine efficacy mediated by nonneutralizing antibody effector functions. Proc Natl Acad Sci U S A. 2018 Jun 12;115(24):6267–6272. pmid:29712861 View Article PubMed/NCBI Google Scholar 13. Katzelnick LC, Gresh L, Halloran ME, Mercado JC, Kuan G, Gordon A, et al. Antibody-dependent enhancement of severe dengue disease in humans. 2017 Nov 17;358(6365):929–932. pmid:29097492 View Article PubMed/NCBI Google Scholar 14. Jennewein MF, Alter G. The Immunoregulatory Roles of Antibody Glycosylation. Trends Immunol. 2017 May;38(5):358–372. pmid:28385520. View Article PubMed/NCBI Google Scholar 15. Palmeira P, Quinello C, Silveira-Lessa AL, Zago CA, Carneiro-Sampaio M (2012) IgG placental transfer in healthy and pathological pregnancies. Clin Dev Immunol. 2012;2012: 985646. pmid:22235228; PubMed Central PMCID: PMC3251916. View Article PubMed/NCBI Google Scholar 16. Jennewein MF, Goldfarb I, Dolatshahi S, Cosgrove C, Noelette FJ, Krykbaeva M, et al. Fc Glycan-Mediated Regulation of Placental Antibody Transfer. Cell. 2019 Jun 27;178(1):202–215.e14. pmid:31204102; PubMed Central PMCID: PMC6741440. View Article PubMed/NCBI Google Scholar 17. Martinez DR, Fong Y, Li SH, Yang F, Jennewein MF, Weiner JA, et al. Fc Characteristics Mediate Selective Placental Transfer of IgG in HIV-Infected Women. Cell. 2019 Jun 27;178(1):190–201.e11. pmid:31204101; PubMed Central PMCID: PMC6727200. View Article PubMed/NCBI Google Scholar 18. Martinez DR, Fouda GG, Peng X, Ackerman ME, Permar SR. Noncanonical placental Fc receptors: What is their role in modulating transplacental transfer of maternal IgG? PLoS Pathog. 2018 Aug 30;14(8):e1007161. pmid:30161231; PubMed Central PMCID: PMC6117057. View Article PubMed/NCBI Google Scholar 19. Johansen FE, Kaetzel CS. Regulation of the polymeric immunoglobulin receptor and IgA transport: new advances in environmental factors that stimulate pIgR expression and its role in mucosal immunity. Mucosal Immunol. 2011 Nov;4(6):598–602. pmid:21956244; PubMed Central PMCID: PMC3196803. View Article PubMed/NCBI Google Scholar 20. Johansen FE, Braathen R, Brandtzaeg P. Role of J chain in secretory immunoglobulin formation. Scand J Immunol. 2000 Sep;52(3): 240–248. pmid:10972899 View Article PubMed/NCBI Google Scholar 21. Johansen F-E, Braathen R, Brandtzaeg P. The J Chain Is Essential for Polymeric Ig Receptor-Mediated Epithelial Transport of IgA. J Immunol. 2001 Nov 1;167(9):5185–5192. pmid:11673531. View Article PubMed/NCBI Google Scholar 22. Aleyd E, Heineke MH, van Egmond M. The era of the immunoglobulin A Fc receptor FcalphaRI; its function and potential as target in disease. Immunol Rev. 2015 Nov;268(1):123–138. pmid:26497517. View Article PubMed/NCBI Google Scholar 23. Hansen IS, Baeten DLP, den Dunnen J. The inflammatory function of human IgA. Cell Mol Life Sci. 2019 Mar;76(6):1041–1055. pmid:30498997; PubMed Central PMCID: PMC6513800 View Article PubMed/NCBI Google Scholar 24. Bonner A, Furtado PB, Almogren A, Kerr MA, Perkins SJ (2008) Implications of the near-planar solution structure of human myeloma dimeric IgA1 for mucosal immunity and IgA nephropathy. J Immunol. 2008 Jan 15;180(2):1008–1018. pmid:18178841. View Article PubMed/NCBI Google Scholar 25. Bakema JE, van Egmond M. Immunoglobulin A: A next generation of therapeutic antibodies? mAbs. 2011 Jul 1;3(4): 352–361. pmid:21691145 View Article PubMed/NCBI Google Scholar 26. Stewart WW, Kerr MA. The specificity of the human neutrophil IgA receptor (Fc alpha R) determined by measurement of chemiluminescence induced by serum or secretory IgA1 or IgA2. Immunology. 1990 Nov;71(3): 328–334. pmid:2269470; PubMed Central PMCID: PMC1384427. View Article PubMed/NCBI Google Scholar 27. Gorter A, Hiemstra PS, Leijh PC, van der Sluys ME, van den Barselaar MT, van Es LA, et al. IgA- and secretory IgA-opsonized S. aureus induce a respiratory burst and phagocytosis by polymorphonuclear leucocytes. Immunology. 1987 Jul; 61(3): 303–309. pmid:3610212; PubMed Central PMCID: PMC1453393. View Article PubMed/NCBI Google Scholar 28. Fadlallah J, El Kafsi H, Sterlin D, Juste C, Parizot C, Dorham, K, et al. Microbial ecology perturbation in human IgA deficiency. Sci Transl Med. 2018 May 2;10(439): eaan1217. pmid:29720448 View Article PubMed/NCBI Google Scholar 29. Woof JM. The human IgA-Fc alpha receptor interaction and its blockade by streptococcal IgA-binding proteins. Biochem Soc Trans. 2002 Aug;30(4):491–494. pmid:12196121. View Article PubMed/NCBI Google Scholar 30. Kett K, Brandtzaeg P, Radl J, Haaijman JJ. Different subclass distribution of IgA-producing cells in human lymphoid organs and various secretory tissues. J Immunol. 1986 May 15;136(10):3631–3635. pmid:3517160. View Article PubMed/NCBI Google Scholar 31. Mattu TS, Pleass RJ, Willis AC, Kilian M, Wormald MR, Lellouch AC, et al. The glycosylation and structure of human serum IgA1, Fab, and Fc regions and the role of N-glycosylation on Fcα receptor interactions. J Biol Chem. 1998 Jan 23;273(4):2260–2272. pmid:9442070. View Article PubMed/NCBI Google Scholar 32. Delacroix DL, Dive C, Rambaud JC, Vaerman JP. IgA subclasses in various secretions and in serum. Immunology. 1982 Oct;47(2):383–375. pmid:7118169; PubMed Central PMCID: PMC1555453. View Article PubMed/NCBI Google Scholar 33. Nakajima A, Vogelzang A, Maruya M, Miyajima M, Murata M, Son A, et al. IgA regulates the composition and metabolic function of gut microbiota by promoting symbiosis between bacteria. J Exp Med. 2018 Aug 6;215(8):2019–2034. pmid:30042191; PubMed Central PMCID: PMC6080902. View Article PubMed/NCBI Google Scholar 34. Rochereau N, Drocourt D, Perouzel E, Pavot V, Redelinghuys P, et al. Dectin-1 is essential for reverse transcytosis of glycosylated SIgA-antigen complexes by intestinal M cells. PLoS Biol. 2013 Sep;11(9):e1001658. pmid:24068891; PubMed Central PMCID: PMC3775721. View Article PubMed/NCBI Google Scholar 35. Mathias A, Corthesy B. Recognition of gram-positive intestinal bacteria by hybridoma- and colostrum-derived secretory immunoglobulin A is mediated by carbohydrates. J Biol Chem. 2011 May 13;286(19):17239–17247. pmid:21454510; PMCID: PMC3089566. View Article PubMed/NCBI Google Scholar 36. Maurer MA, Meyer L, Bianchi M, Turner HL, Le NPL, et al. Glycosylation of Human IgA Directly Inhibits Influenza A and Other Sialic-Acid-Binding Viruses. Cell Rep. 2018 Apr 3;23(1):90–99. pmid:29617676; PMCID: PMC5905402. View Article PubMed/NCBI Google Scholar 37. Graziano VR, Wei J, Wilen CB (2019) Norovirus Attachment and Entry. Viruses. 2019 May 30;11(6). pii: E495. pmid:31151248; PMCID: PMC6630345. View Article PubMed/NCBI Google Scholar 38. Stencel-Baerenwald JE, Reiss K, Reiter DM, Stehle T, Dermody TS. The sweet spot: defining virus-sialic acid interactions. Nat Rev Microbiol. 2014 Nov;12(11):739–749. pmid:25263223; PMCID: PMC4791167. View Article PubMed/NCBI Google Scholar 39. Vono M, Eberhardt CS, Auderset F, Mastelic-Gavillet B, Lemeille S, Christensen D, et al. Maternal Antibodies Inhibit Neonatal and Infant Responses to Vaccination by Shaping the Early-Life B Cell Repertoire within Germinal Centers. Cell Rep. 2019 Aug 13;28(7):1773–1784.e5. pmid:31412246. View Article PubMed/NCBI Google Scholar 40. Voysey M, Kelly DF, Fanshawe TR, Sadarangani M, O'Brien KL, Perera R, et al. The Influence of Maternally Derived Antibody and Infant Age at Vaccination on Infant Vaccine Responses: An Individual Participant Meta-analysis. JAMA Pediatr. 2017 Jul 1;171(7):637–646. pmid:28505244; PMCID: PMC5710349. View Article PubMed/NCBI Google Scholar 41. Kim D, Huey D, Oglesbee M, Niewiesk S. Insights into the regulatory mechanism controlling the inhibition of vaccine-induced seroconversion by maternal antibodies. 2011 Jun 9;117(23):6143–51. pmid:21357766; PMCID: PMC3122939. View Article PubMed/NCBI Google Scholar 42. Heyman B, Nose M, Weigle WO. Carbohydrate chains on IgG2b: a requirement for efficient feedback immunosuppression. J Immunol. 1985 Jun;134(6):4018–4023. pmid:3872908. View Article PubMed/NCBI Google Scholar 43. Heyman B, Wigzell H. Immunoregulation by monoclonal sheep erythrocyte-specific IgG antibodies: suppression is correlated to level of antigen binding and not to isotype. J Immunol. 1984 Mar;132(3):1136–1143. pmid:6363534. View Article PubMed/NCBI Google Scholar 44. Chan J, Nirwati H, Triasih R, Bogdanovic-Sakran N, Soenarto Y, Hakimi M, et al. Maternal antibodies to rotavirus: could they interfere with live rotavirus vaccines in developing countries? Vaccine. 2011 Feb 1;29(6):1242–1247. pmid:21147127. View Article PubMed/NCBI Google Scholar 45. Plotkin SA, Katz M, Brown RE, Pagano JS. Oral poliovirus vaccination in newborn African infants. The inhibitory effect of breast feeding. Am J Dis Child. 1966 Jan;111(1):27–30. pmid:5900282. View Article PubMed/NCBI Google Scholar 46. Ruiz-Palacios GM, Perez-Schael I, Velazquez FR, Abate H, Breuer T, Clemens SC, et al. Safety and efficacy of an attenuated vaccine against severe rotavirus gastroenteritis. N Engl J Med. 2006 Jan 5;354(1):11–22. pmid:16394298. View Article PubMed/NCBI Google Scholar 47. Vesikari T, Uhari M, Renko M, Hemming M, Salminen M, et al. Impact and effectiveness of RotaTeq(R) vaccine based on 3 years of surveillance following introduction of a rotavirus immunization program in Finland. Pediatr Infect Dis J. 2013 Dec;32(12):1365–1373. pmid:24051998. View Article PubMed/NCBI Google Scholar 48. Moon SS, Tate JE, Ray P, Dennehy PH, Archary D, Coutsoudis A, et al. Differential profiles and inhibitory effect on rotavirus vaccines of nonantibody components in breast milk from mothers in developing and developed countries. Pediatr Infect Dis J. 2013 Aug;32(8):863–870. pmid:23584581; PMCID: PMC4610365. View Article PubMed/NCBI Google Scholar 49. Moon SS, Wang Y, Shane AL, Nguyen T, Ray P, Dennehy P, et al. Inhibitory effect of breast milk on infectivity of live oral rotavirus vaccines. Pediatr Infect Dis J. 2010 Oct;29(10):919–23. pmid:20442687; PMCID: PMC3704726. View Article PubMed/NCBI Google Scholar 50. Koch MA, Reiner GL, Lugo KA, Kreuk LS, Stanbery AG, Ansaldo E, et al. Maternal IgG and IgA Antibodies Dampen Mucosal T Helper Cell Responses in Early Life. Cell. 2016 May 5;165(4):827–41. pmid:27153495; PMCID: PMC4866587. View Article PubMed/NCBI Google Scholar 51. Zhang Y, Meyer-Hermann M, George LA, Figge MT, Khan M, et al. Germinal center B cells govern their own fate via antibody feedback. J Exp Med. 2013 Mar 11;210(3):457–464. pmid:23420879; PMCID: PMC3600904. View Article PubMed/NCBI Google Scholar 52. Garg AK, Desikan R, Dixit NM. Preferential Presentation of High-Affinity Immune Complexes in Germinal Centers Can Explain How Passive Immunization Improves the Humoral Response. Cell Rep. 2019 Dec 17;29(12):3946–3957.e5. pmid:31851925. View Article PubMed/NCBI Google Scholar 53. Heesters Balthasar A, Chatterjee P, Kim Y-A, Gonzalez Santiago F, Kuligowski Michael P, et al. Endocytosis and Recycling of Immune Complexes by Follicular Dendritic Cells Enhances B Cell Antigen Binding and Activation. Immunity. 2013 Jun 27;38(6):1164–75. pmid:23770227; PMCID: PMC3773956. View Article PubMed/NCBI Google Scholar 54. Lofano G, Gorman MJ, Yousif AS, Yu W-H, Fox JM, Dugast AS, et al. Antigen-specific antibody Fc glycosylation enhances humoral immunity via the recruitment of complement. Sci Immunol. 2018 Aug 17;3(26). pii: eaat7796. pmid:30120121; PMCID: PMC6298214. View Article PubMed/NCBI Google Scholar 55. Mahan AE, Jennewein MF, Suscovich T, Dionne K, Tedesco J, et al. Antigen-Specific Antibody Glycosylation Is Regulated via Vaccination. PLoS Pathog. 2016 Mar 16;12(3):e1005456. pmid:26982805; PMCID: PMC4794126. View Article PubMed/NCBI Google Scholar
|
|
Scooped by
Gilbert C FAURE
November 17, 2020 11:49 AM
|
Various new technologies have been applied for developing vaccines against various animal diseases. Virus-like particle (VLP) vaccine technology was used for manufacturing the porcine circovirus type 2 and RNA particle vaccines based on an alphavirus ...
|
|
Scooped by
Gilbert C FAURE
October 8, 2020 4:09 AM
|
KEY POINTS Rectal balloon collections minimize blood contamination of mucosal Ig. Rectal balloon collections require ring lubrication to reduce blood contamination. Rectal balloon collection is quick and well tolerated in humans. Abstract Measurements of IgG and IgA in human rectal secretions are used to evaluate the Abs elicited by HIV vaccines or the bioaccumulation following immunoprophylaxis at the sites of HIV exposure. To improve sampling methods and tolerability of the procedure, we optimized a balloon device (OriCol) for rectal microbiome sampling requiring 10 second inflation and compared this method to a 5 minute collection using sponges. Lubrication of the device did not interfere with IgG, IgA, or hemoglobin ELISA. Lubricated OriCols inflated to 30 cc minimized hemoglobin contamination (<4.68 ng/ml) compared with collections with two sponge types (Weck-Cel: 267.2 ng/ml, p < 0.0001; and Merocel: 59.38 ng/ml, p = 0.003). Median human serum albumin for OriCols was 14.9 μg/ml, whereas Merocels and Weck-Cels were 28.57 μg/ml (p = 0.0005) and 106.2 μg/ml (p = 0.0002), respectively. Consistent with reduced systemic contamination, the median IgG measured in OriCol-collected rectal secretions (986 ng) was lower than secretions from sponges (Weck-Cel: 8588 ng, p < 0.0001; Merocel: 2509 ng, p = 0.0389). The median IgA yield of samples using the OriCol method (75,253 ng) was comparable to that using Merocel (71,672 ng; p = 0.6942) but significantly higher than Weck-Cel sponges (16,173 ng, p = 0.0336). Median recovery volumes for OriCols were 800 μl, whereas Merocels and Weck-Cels were 615 μl (p = 0.0010) and 655 μl (p = 0.0113), respectively. The balloon device was acceptable among 23 participants, as 85.1% experiencing their first collection ranked it as “seven: acceptable – a lot” or “six: acceptable – somewhat” in a seven-point Likert scale. Therefore, lubricated OriCols inflated to 30 cc allowed for a rapid, well-tolerated, blood-free collection of human rectal secretions. Footnotes This work was supported by the National Institutes of Health/National Institute of Allergy and Infectious Diseases (NIAID) under award numbers UM1AI068618 (to M.J.M.), UM1AI069481 (to M.J.M.), U19AI128914 (to M.J.M.), and P30AI027757. The phase 1 study was also supported by NIAID Grants UM1AI068614 and UM1AI068635. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Abbreviations used in this article: AFAB assigned female at birth AMAB assigned male at birth Hb hemoglobin HSA human serum albumin IQR interquartile range PI1 protease inhibitors type I RAI receptive anal intercourse. Received March 24, 2020. Accepted August 18, 2020. Copyright © 2020 by The American Association of Immunologists, Inc.
|