Your new post is loading...

|
Scooped by
?
Today, 12:48 PM
|
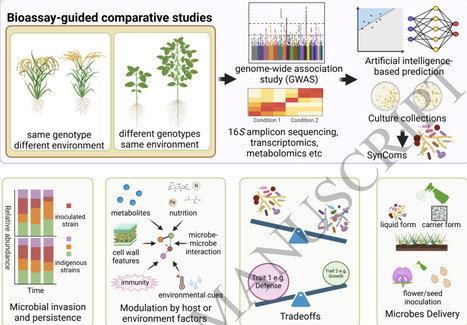
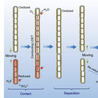
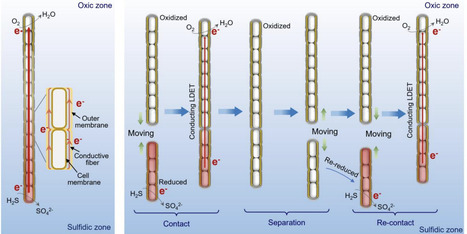
Filamentous cable bacteria are capable of centimeter-scale long-distance electron transport and play crucial roles in the biogeochemistry of aquatic sediments. However, the mechanisms underlying long-distance electron transport remain incompletely understood. This study reports dynamic contacts between separate filaments of cable bacteria, enabling them to relay electrons between sulfidic and oxic zones. Video microscopy of motile filaments in a microchamber slide setup revealed that some filaments did not fully bridge the gap between the sulfidic and oxic zone, but made transient contact with each other. Contacts were always end-to-end and often occurred repeatedly, in which filaments always followed the same trajectory back and forth. The contact frequency gradually increased over the first 20 days, and then declined afterwards. About 5.5% of cable bacterium filaments were observed to engage in contact events during a 2-hour observation window on day 20. Confocal microscopy confirmed the presence of extracellular polymer substance trails between filaments, which appear to guide consecutive end-to-end contacts. In situ Raman spectroscopy showed that connections enabled redox continuity between reduced and oxidized filaments, thus suggesting inter-filament electron transfer during physical contact. This inter-filament electron transport represents a novel type of microbial cooperation, and appears to be a strategy for establishing optimal connections between spatially separated electron donors and acceptors in a dynamic sedimentary environment.
|
|
Scooped by
?
Today, 11:25 AM
|
Droplet microfluidics is a rapidly evolving technology enabling precise control and manipulation of small-volume droplets, typically ranging from picoliters to nanoliters, offering important potential for biomedical applications. By generating highly uniform droplets with size variation below 5% and at high frequencies exceeding 10,000 droplets per second using techniques such as flow focusing, this approach facilitates high-throughput experimentation with minimal reagent consumption. These features make droplet microfluidics invaluable for single-cell analysis, drug screening, and disease diagnostics. Recent advancements in integrating droplet microfluidics with biological and clinical workflows have expanded possibilities for personalized medicine, early disease detection, and high-resolution cellular assays. This review provides an overview of recent progress in droplet microfluidics, focusing on key techniques for droplet generation, manipulation, and detection. It explores their applications in cutting-edge biomedical research, including single-cell analysis, 3-dimensional cell culture, drug development, and cancer research. Additionally, we discuss current challenges, such as improving reproducibility, scalability, and system integration, and outline promising future directions to fully realize the potential of droplet microfluidics in biomedicine.
|
|
Scooped by
?
August 28, 11:18 PM
|
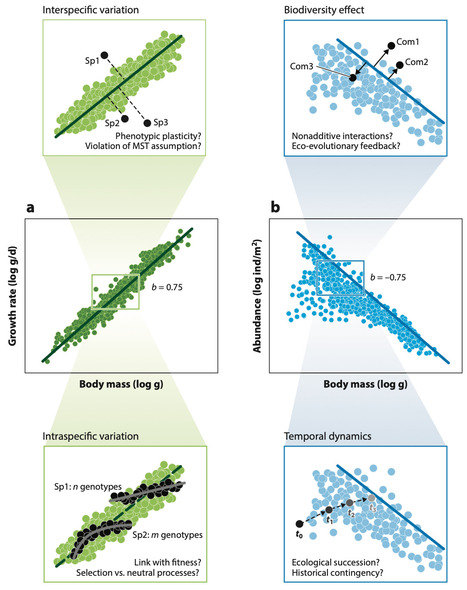
Understanding the relationships between organism traits and ecosystem processes is crucial for advancing ecological theory and predicting ecosystem responses to environmental change. Fundamental biological functions—metabolism, growth, reproduction—scale predictably with organism size across diverse taxa. Moreover, scaling relationships are pervasive across organizational levels, governing population density, competitive interactions, and trophic food webs. This review synthesizes theoretical and empirical evidence linking organism size to ecosystem processes, highlighting the significance of allometric scaling laws, including the metabolic scaling theory and the energy equivalence rule. It discusses deviations from these predictions due to environmental heterogeneity, biotic interactions, and evolutionary dynamics. By exploring the role of size in modulating functional traits and ecosystem-level outcomes, we emphasize the need for integrated approaches to refine predictive models. This review underscores the centrality of size as a key driver of ecological processes and its implications for biodiversity conservation.
|
|
Scooped by
?
August 28, 10:55 PM
|
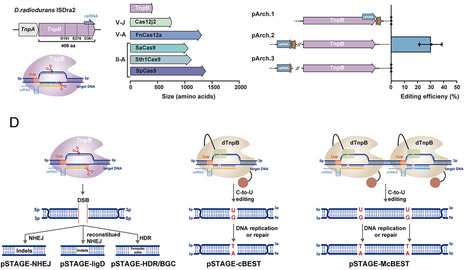
Streptomyces are naturally endowed with the capacity to produce a wide array of natural products with biomedical and biotechnological value. They have garnered great interest in synthetic biology applications given the abundance of uncharacterized biosynthetic gene clusters (BGCs). However, progress has been hindered by the limited availability of genetic tools for manipulating these bacteria. Several representative CRISPR-Cas systems have been established in Streptomyces to streamline experimental workflows and improve editing efficiency. Nevertheless, their broader applicability has been constrained by issues such as nuclease activity-related cytotoxicity and the large size of effector proteins. To address these challenges, we present Streptomyces-compatible TnpB-assisted genome editing (STAGE), a genetic toolkit based on ISDra2 TnpB, which is approximately one-third the size of Cas9 and enables precise, site-specific gene editing. We demonstrated that STAGE introduces genetic mutations with high efficiency and minimal off-target effects in two industrially important Streptomyces strains. Building on this platform, we developed STAGE-cBEST and STAGE-McBEST, enabling single and multiplexed C·G-to-T·A base editing, respectively, with editing efficiencies exceeding 75%. To further enhance performance, we engineered the ISDra2 TnpB system using an AI-assisted protein engineering framework, resulting in two variants that achieve nearly 100% genome editing efficiency. Additionally, through sequence homology analysis, we identified a TnpB ortholog from the same biological origin of ISDra2 TnpB, which also functions effectively as a gene editing tool. Our study establishes STAGE as a highly precise, programmable, and versatile genome editing platform for Streptomyces, paving the way for advanced genetic manipulation and synthetic biology applications in these industrially important bacteria.
|
|
Scooped by
?
August 28, 10:34 PM
|
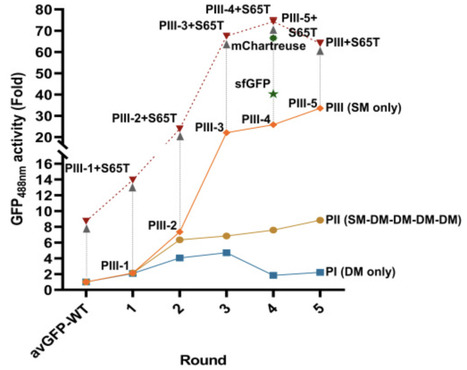
Deep learning has rapidly emerged as a promising toolkit for protein optimization, yet its success remains limited, particularly in the realm of activity. Moreover, most algorithms lack rigorous iterative evaluation, a crucial aspect of protein engineering exemplified by classical directed evolution. This study introduces DeepDE, a robust iterative deep learning-guided algorithm leveraging triple mutants as building blocks and a compact library of ∼1,000 mutants for training. Triple mutants allow for the exploration of a much greater sequence space compared to single or double mutants in each iteration. When applied to GFP from Aequorea victoria, DeepDE achieved a remarkable 74.3-fold increase in activity over four rounds of evolution, far surpassing the benchmark superfolder GFP. Our study suggests that limited screening involving experimentally affordable ∼1,000 variants significantly enhances the performance of DeepDE, likely by mitigating the constraints imposed by the intractable data sparsity problem in protein engineering.
|
|
Scooped by
?
August 28, 4:40 PM
|
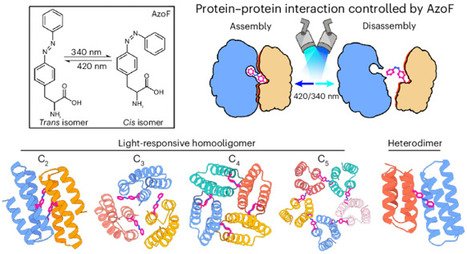
Light-responsive proteins play an essential role in all domains of life by sensing and responding to environmental light signals. However, the de novo design of light-responsive proteins with precisely defined structures and reversible responsive behaviors is an unmet challenge. Here we describe a computational approach to design protein–protein interactions regulated by non-canonical amino acids, focusing on the light-responsive phenylalanine-4′-azobenzene (AzoF). Using this approach, we designed light-responsive cyclic homo-oligomers and heterodimers, which only assemble in AzoF’s trans configuration and disassemble when AzoF photoisomerizes to the cis configuration. Biophysical characterization confirms the light-responsive assembly and disassembly of these complexes, and the crystal structures match the design models with atomic accuracy. We demonstrate the applicability of these light-responsive proteins in constructing light-responsive hydrogels and engineering synthetic ligand receptors to optocontrol cell signalling in mammalian cells. Our approach opens avenues for designing environmentally responsive protein structures and broadens the toolkit for optogenetics and optochemistry. Light-responsive proteins hold significant potential in biotechnology but naturally occurring examples are scarce and limited in diversity. Now a totally de novo computational approach has been developed to design light-responsive proteins with diverse architectures by precisely incorporating the non-canonical amino acid phenylalanine-4′-azobenzene (AzoF), which is responsive to light.
|
|
Scooped by
?
August 28, 4:03 PM
|
Developing efficient and simplified tools for multiplexed genome editing remains challenging due to limitations in precursor CRISPR RNA (pre-crRNA) processing and reliance on additional RNA-based regulatory components. Cas12i.3, a small RNA-guided nuclease, reportedly lacks pre-crRNA processing ability, restricting its multiplexing capability. Here, we engineered Cas12i.3 by optimizing CRISPR RNA (crRNA) design, codon usage, and exonuclease fusion, generating initial optimized Cas12i (IOCas12i) system. Further rational design and amino acid mutations yielded the highly efficient enhanced optimized Cas12i (EOCas12i) systems, EOCas12i–Combo1 and EOCas12i–Combo2, exhibiting 2.5- to 22.8-fold and 3.0- to 60.0-fold editing efficiencies relative to wild-type Cas12i.3, comparable to Streptococcus pyogenes Cas9 (SpCas9) and Lachnospiraceae bacterium Cas12a (LbCas12a). Additionally, they exhibited high specificity and produced longer insertions and deletions (indels) that may facilitate gene knockout. Notably, both variants enabled efficient multiplexed editing of up to 30 targets using compact crRNA arrays. These advancements position EOCas12i–Combo1 and EOCas12i–Combo2 as promising platforms for multiplexed genome editing applications.
|
|
Scooped by
?
August 28, 3:53 PM
|
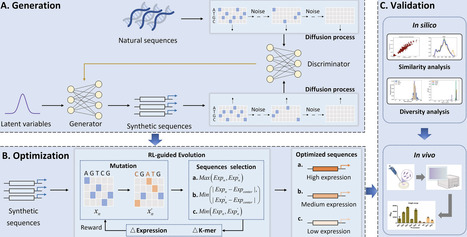
Promoters are core elements in regulating gene expression. The design and optimization of functional promoters is crucial for enhancing metabolic pathway construction and advancing gene therapy. Deep learning-based methods have shown great potential in promoter design. However, existing studies mainly focus on designing strong promoters, neglecting the practical need for promoters with varying regulatory intensities. Here, we propose a novel promoter design method, PromoDGDE, to design promoters with desirable expression levels and apply it to the promoter design of Escherichia coli and Saccharomyces cerevisiae. It uses Diffusion-GAN to learn the feature distribution of natural sequences and generate new promoters. Then, reinforcement learning and evolutionary algorithms are combined to dynamically optimize the synthetic sequences. In silico analyze results demonstrate that PromoDGDE outperforms existing methods, generating promoters that not only possess biological significance but also achieve the intended function. In vivo experiment results demonstrate that the synthetic promoters exhibit expression activity, with over 60% of the sequences showing the expected regulatory effects. These results confirm the practical effectiveness of PromoDGDE and demonstrate its ability to provide an efficient and flexible solution for complex design needs in synthetic biology.
|
|
Scooped by
?
August 28, 12:54 PM
|
Rational engineering strategies that seek to harness the remarkable diversity of microbial metabolism can be limited by incomplete biological knowledge. As described here, a novel approach to address this challenge involved replacing a native pathway for degrading lignin-derived aromatic compounds via ortho cleavage of protocatechuate in Acinetobacter baylyi ADP1 with a foreign meta-cleavage pathway that uses different enzymes, metabolites, and redox carriers. This alteration may improve lignin valorization and coordinate catabolism with bioproduction strategies. When a 14-kbp region of foreign DNA was inserted in the chromosome, the heterologous genes failed to confer growth on target substrates. Regional gene dosage was increased using a synthetic DNA fragment to promote recombination, and higher copy number enabled growth. During adaptive laboratory evolution, compensatory mutations arose that permit growth with one copy of the foreign genes. This complex metabolic remodeling was accomplished without assumptions about the impediments that initially prevented growth. To understand the changes that emerged, a novel transformation assay identified a combination of mutations sufficient for the new phenotype. Three unexpected changes were revealed: loss of one foreign enzyme, loss of one native enzyme, and loss of a two-component transcriptional regulatory system. This study establishes that large multicopy tandem arrays of poorly adapted pathway genes can confer new functions and improve understanding of metabolism.
|
|
Scooped by
?
August 28, 12:41 PM
|
New microbial hosts with superior phenotypes, such as fast growth, are attractive for research and biotechnology, but often lack systematic evaluation of functional genetic parts. A reference set of working plasmids would increase reproducibility and encourage use of these hosts. Here, we use the POSSUM toolkit, a collection of 23 origins-of-replication and 6 antibiotic markers, to identify functional genetic parts for strains of Vibrio natriegens. We applied this to the wild-type strain ATCC 14048 and an engineered variant NBx CyCloneTM, evaluating 414 combinations of origins of replication and antibiotic selection conditions. We show that both strains support five replicons (pNG2, pSa, pSC101ts, p15A, and RSF1010) with NBx CyCloneTM supporting an extra replicon RK2. The assay can be performed in under a week and is compatible with multiple DNA delivery methods. This work demonstrates the feasibility of rapidly establishing reference information to accelerate the adoption of new microbial hosts.
|
|
Scooped by
?
August 28, 12:16 PM
|
Promoters are DNA sequences that help to initiate transcription. Point mutations can create de-novo promoters, which can consequently transcribe inactive genes or create novel transcripts. We know little about how de novo promoters emerge in genomic DNA, especially compared to random DNA that has never been subjected to selection. Here, we assayed the promoter activity of 17,129 random, synthetic DNA sequences and 91,866 E. coli genomic DNA sequences. Genomic DNA encodes ~1.3 times more promoters than random DNA. We then studied 584,573 point mutations in 225 random and 60 genomic sequences, and asked how they cause the emergence of de-novo promoters. We find that de-novo promoters emerge ~3 times more readily from random DNA than from genomic DNA. The reason is that the genome contains fewer proto-binding sites for transcriptional activators than random DNA. Our work shows that the evolutionary history of a DNA sequence introduces substantial biases in its evolutionary potential, especially in the likelihood that mutations create new and potentially adaptive transcripts.
|
|
Scooped by
?
August 28, 12:02 PM
|
Oscillations are fundamental to biological timekeeping and organization, yet understanding how their complex temporal dynamics emerge from underlying molecular interactions remains a significant challenge. In vitro reconstitution offers a powerful bottom-up approach to dissect the minimal components, interactions, and parameters required to generate these rhythmic behaviors. Biochemical reconstruction of minimal oscillators outside of their native cellular contexts allows the direct interrogation of the biochemical, biophysical, and systems-level properties that govern oscillatory dynamics and unravel the governing fundamental design principles. In this review, we summarize the theoretical foundations of biological oscillators and outline the major experimental challenges associated with their in vitro reconstitution. We highlight recent advances in the reconstitution of diverse oscillator types, including the cyanobacterial circadian clock, the Min system from Escherichia coli, and synthetic genetic oscillators such as the repressilator. These case studies illustrate how reconstitution efforts have yielded key mechanistic insights and driven technological innovation. We conclude by exploring emerging tools and future directions that promise to overcome current limitations and broaden the applicability of oscillator reconstitution–both to additional biological systems and to a wider range of scientific questions.
|
|
Scooped by
?
August 28, 11:56 AM
|
The determination of RNA secondary structure (RSS) could help understand RNA’s functional mechanisms, guiding the design of RNA-based therapeutics, and advancing synthetic biology applications. However, traditional methods such as NMR for determining RSS are typically time-consuming and labor-intensive. As a result, the accurate prediction of RSS remains a fundamental yet unmet need in RNA research. Various deep learning (DL)-based methods achieved improved accuracy over thermodynamic-based methods. However, the over-parameterization nature of DL makes these methods prone to overfitting and thus limits their generalizability. Meanwhile, the inconsistency of RSS predictions between these methods further aggravated the crisis of generalizability. Here, we propose TrioFold to achieve enhanced generalizability of RSS prediction by integrating base-pairing clues learned from both thermodynamic- and DL-based methods by ensemble learning and convolutional block attention mechanism. TrioFold achieves higher accuracy in intra-family predictions and enhanced generalizability in inter-family and cross-RNA-types predictions. Additionally, we have developed an online webserver equipped with widely used RSS prediction algorithms and analysis tools, providing an accessible platform for the RNA research community. This study demonstrated new opportunities to improve generalizability for RSS predictions by efficient ensemble learning of base-pairing clues learned from both thermodynamic- and DL-based algorithms.
|
|
|
Scooped by
?
Today, 12:42 PM
|
Bacterial genome exploration and outbreak analysis rely heavily on robust whole-genome sequencing and bioinformatics analysis. Widely-used genomic methods, such as genotyping and detection of genetic markers demand high sequencing accuracy and precise genome assembly for reliable results. Methods To assess the utility of nanopore sequencing for genotyping highly pathogenic bacteria with low mutation rates, we sequenced six reference strains using Oxford Nanopore Technologies (ONT) R10.4.1 chemistry and Illumina and evaluated different assembly strategies. The publicly available RefSeq assemblies were chosen as the ground truth. Publicly available sequencing data from key foodborne and public-health-related bacterial pathogens were examined to provide a broader context for the analysis. While for Bacillus (Ba.) anthracis an almost perfect assembly was achieved, results varied for other species. For Brucella (Br.) spp., the final assemblies comprised five to 46 different nucleotides in comparison to Sanger-sequenced references. For some key foodborne and public-health-related bacterial pathogens (Klebsiella (K.) variicola, Listeria spp., Mycobacterium (M.) tuberculosis, Staphylococcus (Sta.) aureus, and Streptococcus (Str.) pyogenes) perfect genomes were obtained. Enhanced basecalling models have generally improved assembly accuracy, however, for certain species such as Br. abortus, older models have produced higher accuracy. While long-read polishing mainly improves assembly quality with only one round needed, our results indicate that this process may also degrade assembly quality. Overall, 81% of the observed errors in ONT assemblies were located within coding sequences (CDS). Furthermore, we found that methylation caused 6.5% of the errors, and the bacterial methylation-aware medaka polishing model reduced the number of errors linked to methylation. Core-genome Multilocus Sequence Typing (cgMLST) analysis revealed allele differences in Ba. anthracis, Br. abortus, and Francisella (F.) tularensis for some assemblers, although with fewer than five allele differences. In the case of Br. melitensis, some assemblies included five allele differences, whereas for Br. suis the correct cgMLST alleles were observed. Assembling nanopore data from pathogenic bacteria vary in quality across different species and methods. However, errors persist in the final assemblies, including within cgMLST loci, influencing the reliability of outbreak predictions. Nevertheless, specific combinations of existing tools can generate perfect genome assemblies from bacterial ONT sequencing data for outbreak analysis without short-read polishing.
|
|
Scooped by
?
August 28, 11:24 PM
|
Symbiotic associations between plants and microorganisms are crucial to global biogeochemical cycling and ecosystem stability. Mycorrhizal fungi and nitrogen (N2)-fixing bacteria are recognized as the two main groups of microorganisms involved in such symbiotic interactions. They not only constitute the most wide-spread symbiotic microorganisms, but also ensure plants to acquire additional N resources directly from the atmosphere. Although plant-microbial interactions, for example, the performance of AM-plant and rhizobia-legume plant symbioses, have been well studied and reviewed in detail previously, still less information is known about these processes in actinorhizal symbioses. The present review is aimed to summarize current knowledge of the interaction of partners in actinorhizal root symbioses, in particular the signalling processes during establishment of BNF, and the specificity of and dependency on different symbiotic partners in this interactions, based on evolution and distribution in the plant and microbial kingdom. The features of nutrient transfer in these root symbiotic relationships and the significance of actinorhizal symbioses for the performance of plants under environmental stress are discussed and compared with AM and rhizobia-legume symbioses. In addition, research gaps in actinorhizal root symbioses research are identified and future research avenues are suggested.
|
|
Scooped by
?
August 28, 11:11 PM
|
As temperatures rise, plants are expected to shift their ranges to align with their abiotic niches. If plants do not encounter suitable mycorrhizal fungi in new habitats, however, these migrations may fail. We review the literature to describe how arbuscular mycorrhizal (AM) fungi, ectomycorrhizal (EM) fungi, and ericoid mycorrhizal (ErM) fungi currently vary within and beyond host plants’ ranges and how these mycorrhizal fungi shape plant ranges. We introduce a framework that predicts when plants are likely to encounter suitable mycorrhizal mutualists in new habitats. Critically, the probability of beneficial plant–mycorrhizal fungal interactions occurring depends on (a) plants’ specificity to mycorrhizal fungi, (b) abiotic similarity between historic and new ranges, (c) plants’ relatedness to new range plants, (d) geographic distance between historic and new ranges, and (e) alignment of plant and mycorrhizal fungal niches, all of which are affected by mycorrhizal guild. To conclude, we review research frontiers in the field of plant–mycorrhizal fungal interactions.
|
|
Scooped by
?
August 28, 10:49 PM
|
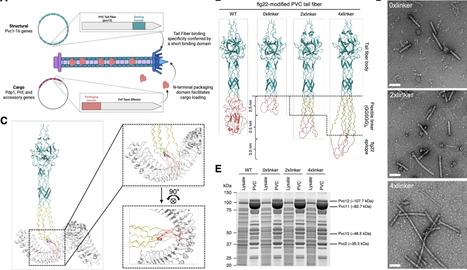
Efficient delivery of functional proteins into plant cells remains a major barrier in plant biotechnology. Extracellular contractile injection systems (eCISs) are phage tail-like nanomachines evolved by bacteria to interface with eukaryotic host cells and deliver protein effectors. The Photorhabdus virulence cassette (PVC), a well-characterized eCIS, naturally targets insect hosts but can be reprogrammed for protein cargo delivery in mammalian systems. Here, we adapted PVCs for targeted delivery to plants by engineering their tail fibers to recognize a natural plant immune receptor, FLAGELLIN SENSITIVE2 (FLS2). We designed a library of FLS2-binding PVC variants and demonstrated efficient loading and delivery of non-native cargoes, including a fluorescent reporter protein and the Cre recombinase. We showed that engineered PVCs can deliver these proteins to Arabidopsis thaliana protoplasts and Nicotiana benthamiana leaf cells with efficiencies up to 40%. We elucidated that the delivery efficiency is correlated with receptor surface density, demonstrating that receptor selection and expression level are key parameters for optimization. This work establishes PVCs as novel, programmable protein delivery nanoparticles for plants, capable of targeting plant membrane receptors and effectively delivering diverse functional proteins. By enabling precise, DNA-free delivery of gene editing proteins, plant-targeted PVCs provide the framework for next-generation genome engineering strategies with broad potential in agricultural nanobiotechnology.
|
|
Scooped by
?
August 28, 10:21 PM
|
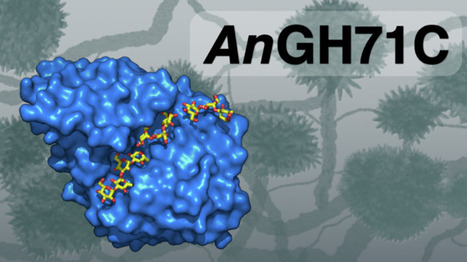
The microbial polysaccharide α-1,3-glucan is an important component of fungal cell walls and dental plaque biofilms, contributing to microbial virulence and biofilm resilience. Glycoside hydrolase family 71 (GH71) includes α-1,3-glucan degrading enzymes which could be exploited for biotechnological applications; however, the family is presently poorly understood. To increase our understanding of GH71, we have performed a phylogenetic analysis of the family and detailed biochemical analysis of two of the five GH71 enzymes encoded by Aspergillus nidulans (AnGH71B and -C). Both are active on soluble α-1,3-glucooligosaccharides but surprisingly only minimally on water-insoluble α-1,3-glucan. Assays on intact and milled A. nidulans biomass indicate that the enzymes act on fungal cell wall glycosidic linkages, likely having roles in cell wall remodelling. Both enzymes utilize an inverting mechanism but differ in specificity and product profiles indicating exo- and endo-like activity for AnGH71B and AnGH71C, respectively. We present the first structure of a GH71 protein, AnGH71C, including structures with carbohydrate ligands. These structures revealed a conserved acidic dyad (DxxE), found to be crucial for activity, and active site water coordination consistent with a classical inverting GH mechanism. This work provides new insights into GH71, highlighting its functional diversity and the enzymes roles in fungal physiology. Mazurkewich et al. report a phylogenetic, biochemical, and structural investigation of GH71 enzymes from Aspergillus nidulans, revealing distinct activities on α-1,3 glucooligosaccharides and roles in fungal cell wall remodeling. Their work uncovers the first GH71 structure with bound ligands, identifies a crucial DxxE catalytic dyad, and highlights the functional diversity of this poorly understood enzyme family.
|
|
Scooped by
?
August 28, 4:19 PM
|
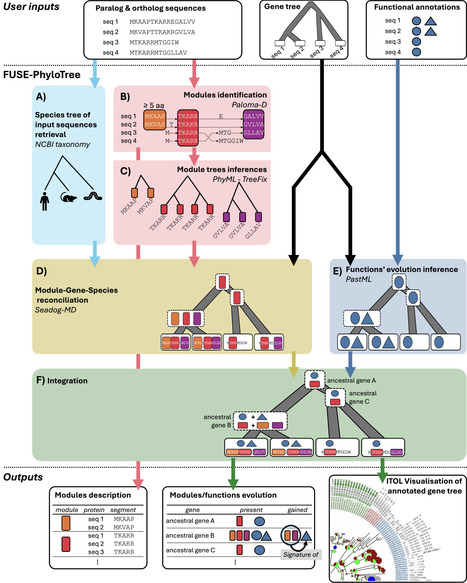
FUSE-PhyloTree is a phylogenomic analysis software for identifying local sequence conservation associated with the different functions of a multi-functional (e.g., paralogous or multi-domain) protein family. FUSE-PhyloTree introduces an original approach that combines advanced sequence analysis with phylogenetic methods. First, local sequence conservation modules within the family are identified using partial local multiple sequence alignment. Next, the evolution of the detected modules and known protein functions is inferred within the family’s phylogenetic tree using three-level phylogenetic reconciliation and ancestral state reconstruction. As a result, FUSE-PhyloTree provides a gene tree annotated with both predicted sequence modules and ancestral gene functions, enabling the association of functions with specific sequence regions based on their co-emergence. FUSE-PhyloTree is provided as Docker and Singularity images including all the required software tools. Images, source code, test data, and documentation are available at https://github.com/OcMalde/fuse-phylotree and
|
|
Scooped by
?
August 28, 3:59 PM
|
Sequence-specific interactions of transcription factors (TFs) with genomic DNA underlie many cellular processes. High-throughput in vitro binding assays coupled with machine learning have made it possible to accurately define such molecular recognition in a biophysically interpretable way for hundreds of TFs across many structural families, providing new avenues for predicting how the sequence preference of a TF is impacted by disease-associated mutations in its DNA binding domain. We developed a method based on a reference-free tetrahedral representation of variation in base preference within a given structural family that can be used to accurately predict the effect of mutations in the protein sequence of the TF. Using the basic helix-loop-helix (bHLH) and homeodomain (HD) families as test cases, our results demonstrate the feasibility of accurately predicting the shifts (ΔΔΔG/RT) in binding free energy associated with TF mutants by leveraging high-quality DNA binding models for sets of homologous wild-type TFs.
|
|
Scooped by
?
August 28, 3:42 PM
|
Metatranscriptomics methods for the skin are hampered by low microbial biomass, contamination with host cells and low RNA stability. In this study, we developed a robust, clinically tractable skin metatranscriptomics workflow that provides high technical reproducibility of profiles, uniform coverage across gene bodies and strong enrichment of microbial mRNAs. Paired application of this protocol with metagenomics to five skin sites in a cohort of 27 healthy adults identifies a notable divergence between transcriptomic and genomic abundances. Specifically, Staphylococcus species and the fungi Malassezia had an outsized contribution to metatranscriptomes at most sites, despite their modest representation in metagenomes. Species-level analysis shows signatures of microbial adaptation to their niches. Gene-level analysis identifies diverse antimicrobial genes transcribed by skin commensals in situ, including several uncharacterized bacteriocins. Correlation of microbial gene expression with organismal abundances uncovers more than 20 genes that putatively mediate interactions between microbes. This work highlights how skin metatranscriptomics identifies active species and microbial functions in situ. Skin metagenomic and metatranscriptomic analysis shows divergence between microbial abundance and activity.
|
|
Scooped by
?
August 28, 12:49 PM
|
Inorganic phosphorus (Pi) is vital for all living organisms, but abnormal Pi levels in water and serum pose significant eutrophication and health risks. This study developed a highly sensitive PhoR-PhoB-based biosensor for ultralow-level Pi detection in environmental and serum samples. The Escherichia coli PhoR-PhoB system comprising the histidine kinase PhoR and the response regulator PhoB functions as a Pi-sensing two-component system. However, the limited dynamic range and suboptimal detection range of the native PhoR-PhoB biosensor restrict its practical application. To address these limitations, the expression levels of PhoR and PhoB were optimized, increasing the dynamic range to 97-fold. Directed evolution on the kinase domain of PhoR yielded the mutant PhoR (L222F/N307S/Q344H), which exhibited a 232-fold dynamic range and a linear detection range of 0.012–0.07 mM. This range covers normal serum Pi levels and meets international wastewater discharge standards. The optimized biosensor was successfully applied to Pi detection in serum and environmental water samples, demonstrating its potential for precise measurement of ultralow Pi levels in diverse environments.
|
|
Scooped by
?
August 28, 12:38 PM
|
Programmable DNA integration using CRISPR-associated transposons (CASTs) offers powerful capabilities for genome engineering. The single effector Cas12k CAST examples evolved from a fixed guide TnpB nuclease protein. Here, we engineer de novo RNA-guided transposition systems, where the single guide RNA effector components are repurposed nuclease-dead TnpB-family proteins. These compact systems mediate high-efficiency guide RNA-directed DNA insertion with preserved orientation control and target immunity, reduced off-site targeting, release of a host factor requirement, and can be paired with an exonuclease domain to mediate cut-and-paste transposition. In this engineered context, the TnpB derivatives show features not predicted from the original enzymes suggesting untapped avenues for improvement. In parallel, we show that mutations at the TniQ-TnsC interface in the Cas12k CAST system selectively attenuate off-site insertions while enhancing on-site activity. These results establish how Cas12 proteins and antecedent TnpB proteins can be engineered for high performance and specificity with guide RNA directed systems.
|
|
Scooped by
?
August 28, 12:07 PM
|
New reference genomes and transcriptomes are increasingly available across the tree of life, opening new avenues to tackle exciting questions. However, there are still challenges associated with annotating genomes and inferring evolutionary processes and with a lack of methodological standardization. Here, we propose a new workflow designed for evolutionary analyses to overcome these challenges, facilitating the detection of recombination suppression and its consequences in terms of rearrangements and transposable element accumulation. To do so, we assemble multiple bioinformatic steps in a single easy-to-use workflow. We combine state-of-the-art tools to detect transposable elements, annotate genomes, infer gene orthology relationships, compute divergence between sequences, infer evolutionary strata (i.e. footprints of stepwise extension of recombination suppression) and their structural rearrangements, and visualise the results. This workflow, called EASYstrata, was applied to reannotate 42 published genomes from Microbotryum fungi. We show in further case examples from a plant and an animal that we recover the same strata as previously described. While this tool was developed with the goal to infer divergence between sex or mating-type chromosomes, it can be applied to any pair of haplotypes whose pattern of divergence is of interest. This workflow will facilitate the study of non-model species for which newly sequenced phased diploid genomes are becoming available.
|
|
Scooped by
?
August 28, 11:59 AM
|
Plants must contend with oxidative stress, a paradoxical phenomenon in which reactive oxygen species (ROS) can cause cellular damage while also serving as key signaling molecules. Environmental stressors, such as drought, salinity, and temperature extremes, promote ROS accumulation, affecting plant growth and productivity. To maintain redox homeostasis, plants rely on antioxidant systems comprising enzymatic defenses, such as superoxide dismutase, catalase, and ascorbate peroxidase, and non-enzymatic molecules, including ascorbate, glutathione, flavonoids, and emerging compounds such as proline and nano-silicon. This review provides an integrated overview of antioxidant responses and their modulation through recent biotechnological advances, emphasizing the role of emerging technologies in advancing our understanding of redox regulation and translating molecular insights into stress-resilient phenotypes. Omics approaches have enabled the identification of redox-related genes, while genome editing tools, particularly those based on clustered regularly interspaced short palindromic repeats (CRISPR) and CRISPR-associated (Cas) proteins, offer opportunities for precise functional manipulation. Artificial intelligence and systems biology are accelerating the discovery of regulatory modules and enabling predictive modeling of antioxidant networks. We also highlight the contribution of synthetic biology to the development of stress-responsive gene circuits and address current regulatory and ethical considerations. Overall, this review aims to provide a comprehensive perspective on molecular, biochemical, and technological strategies to enhance oxidative stress tolerance in plants, thereby contributing to sustainable agriculture and food security in a changing climate.
|