 Your new post is loading...

|
Scooped by
?
Today, 4:58 PM
|
Biomolecular condensates are membraneless compartments involved in a wide range of cellular processes. Despite their fundamental role in the spatiotemporal regulation of cellular functions, tools for precisely manipulating phase-separated condensates remain limited, and effective methods for discovering and functionalizing tunable phase separation modules from natural proteins are lacking. Here we present a rational engineering approach for androgen receptor (AR) and its clinically used drugs to create a chemical genetic platform, ARDrop, enabling condensates formation and dissolution. This platform is applied to a diverse set of proteins to achieve intended cellular functions, ensuring robust and long-lasting functionality through stable liquid-like properties. Our work develops a powerful toolkit for reversible manipulation of condensates that can be used for dissection of complicated cell signaling, laying the foundation for engineering designer condensates for synthetic biology applications. Tools for precisely manipulating phase-separated condensates remain limited. Here authors present an engineered system based on the androgen receptor, where clinically used drugs can be used to control the formation and dissolution of synthetic condensates, subsequently allowing control over various cellular processes.
|
|
Scooped by
?
Today, 4:40 PM
|
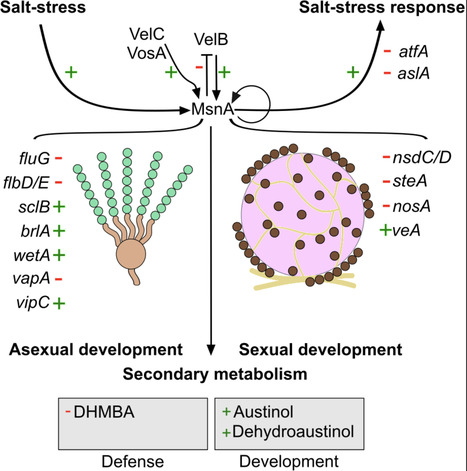
Development and secondary metabolism of the filamentous fungus Aspergillus nidulans are tightly controlled by concerted actions of several master regulator transcription factors (TFs). The connection between fungal development and cellular stress response programs is often elusive. Here we show that the zinc finger TF MsnA, which controls salt-stress response, is a novel major regulator of fungal development. A molecular circuit among MsnA and the velvet domain regulator VelB was discovered, which mutually fosters the actions of both regulatory proteins during development. Chromatin immunoprecipitation coupled with next generation sequencing (ChIP-seq) and gene expression studies have revealed that MsnA controls the expression of several genes encoding key transcriptional regulators of asexual as well as sexual development. The double mutant of msnA with velB showed that both genes share an additive genetic relationship, under normal and salt stress conditions, with each protein to control distinct phenotypical features. In addition, MsnA directly and indirectly affects the synthesis of specific secondary metabolites relevant for fungal defense against other organisms and growth, in addition to salt-stress responses. Moreover, the expression of genes encoding the epigenetic regulators VapA, VipC and LaeA are also directly controlled by MsnA. The VapA-VipC-VapB methyltransferase signal transduction complex promotes asexual differentiation, while the VeA-VelB-LaeA complex balances light response, development and the secondary metabolism of the fungus. MsnA is therefore placed at a novel prominent position of the central regulatory network, which coordinates stress responses with the developmental and metabolic fate of the fungus.
|
|
Scooped by
?
Today, 4:22 PM
|
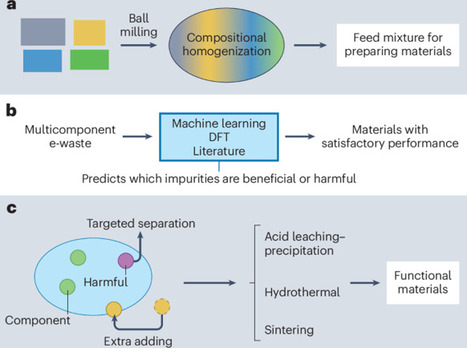
Discarded electronic waste is an environmental burden but holds promise as a sustainable source for manufacturing functional materials such as adsorbents, catalysts and electrodes. Addressing the challenges in re-using electronic waste-derived materials will require using the multi-component waste stream, predictive impurity control and standardized methods of cost assessment.
|
|
Scooped by
?
Today, 12:58 PM
|
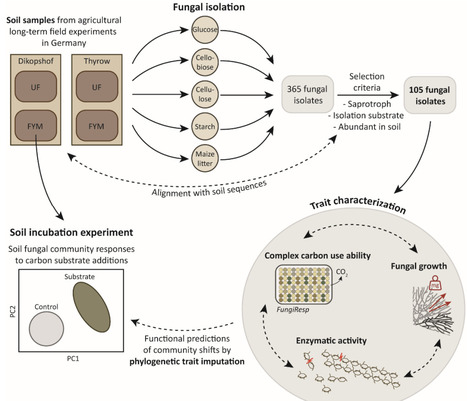
Fungal communities in soil play important roles in decomposition processes and soil organic carbon cycling. These communities are tremendously diverse, making it challenging to assign relevant functions to individual species. Fungal communities may be differentiated at the level of functional guilds; beyond such broad classification we have little delimitation, especially in fungal taxa common in grassland and agricultural soils. To resolve the level of functional similarity in fungal communities and define traits predictive of soil carbon cycling, we characterized fungal isolates abundant in agricultural soils to test the hypotheses that (i) the majority of saprobic soil fungi have the ability to use complex carbon sources, (ii) differences in complex carbon use abilities correlate with fungal enzymatic profiles, following principles of the fungal economics spectrum and (iii) carbon use ability is a predictive trait for fungal community functions. Using specialized growth media, we isolated and characterized 105 isolates and developed a novel FungiResp approach that directly tests fungal activity on complex carbon sources. The largest amount of variance between isolates was explained by differential abilities to use cellulose and starch, with only few phylogenetically distinct fungal clades showing high respiratory activity on these biopolymers. A preference for bacterial necromass was another major distinction among taxa. These key traits correlated with soil fungal community shifts in response to carbon substrate availability. By contrast, enzymatic activity was a poor predictor of fungal carbon use ability, except for correlations in lignin use and laccase activity. The newly established functional trait of carbon use ability offers important insights into diverse fungal communities: Many taxa lack the ability to use complex carbon (on their own), while the most common enzymes analyzed in soil showed little correlation with fungal mineralization potential. The discovery of key functional traits is an important step towards predicting the significance of fungal community shifts for soil carbon cycling.
|
|
Scooped by
?
Today, 12:47 PM
|
Recent advances in deep learning, particularly transformer architectures, have improved computational approaches for biological sequence analysis. Despite these advances, computational models for bacterial promoter prediction have remained limited by small datasets, species-specific training, and binary classification approaches rather than comprehensive annotation frameworks. We present PromoterAtlas, a 1.8M parameter transformer model trained on 9M regulatory sequences from 3,371 gammaproteobacterial species. The model demonstrates recognition of various regulatory elements across different species, including ribosomal binding sites, various types of bacterial promoters, transcription factor binding sites, and terminators. Using this model, we developed a whole-genome promoter annotation tool for Gammaproteobacteria, with various levels of validation that support the predictions of promoters associated with different sigma (σ) factors. Furthermore, we show that the model embeddings encode cross-species evolutionary relationships, clustering promoters by σ factor identity rather than species-specific sequence features. Finally, we show that model embeddings encode regulatory sequence information that enables effective prediction of transcription and translation levels. PromoterAtlas can contribute to our understanding of and ability to engineer bacterial regulatory sequences with potential applications in bacterial biology, synthetic biology, and comparative genomics.
|
|
Scooped by
?
Today, 12:32 PM
|
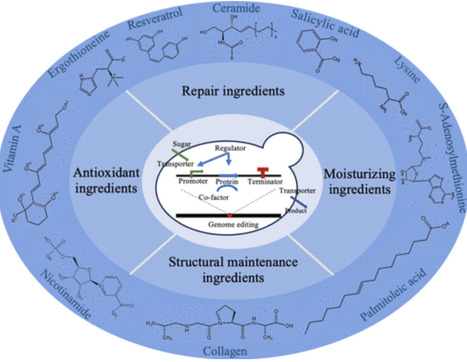
Driven by consumer preferences for safety and environmental protection, the global cosmetics industry has an increasing demand for natural and sustainable ingredients. Saccharomyces cerevisiae has emerged as a powerful platform for the biosynthesis of cosmetic ingredients due to its strong metabolic capacity, genetic operability, and cost-effective production capabilities. This review focuses on the latest advances in S. cerevisiae for the production of high-value cosmetic compounds, including antioxidants, repair agents, moisturizers, and structure-maintaining ingredients. Key strategies, such as genetic and metabolic engineering, pathway modularity, and fermentation optimization, are discussed, demonstrating significant improvements in yield and efficiency. In addition, the integration of artificial intelligence and machine learning in strain design and process control is explored, providing promising solutions to overcome metabolic bottlenecks and scale up production. Despite challenges such as metabolic burden, S. cerevisiae shows great potential for sustainable and scalable biosynthesis of cosmetic ingredients, paving the way for the next generation of biobased cosmetics. This comprehensive review provides valuable insights and technical references for the development of the field of synthetic biology in the cosmetics industry.
|
|
Scooped by
?
Today, 12:08 PM
|
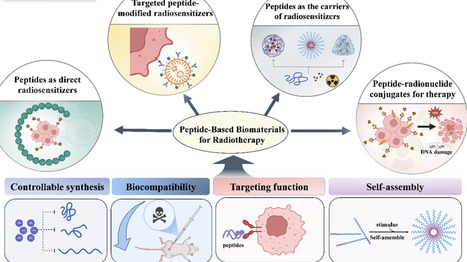
Radiotherapy (RT) is a mainstay therapeutic strategy for cancer; however, increasing radiation damage to tumor tissues while reducing the side effects on healthy tissues remains a great challenge. To address this issue, the use of biomaterials has proven promising. Recently, attention has been paid to peptide-based biomaterials as platforms for enhancing radiotherapeutic efficacy. This review explores peptide-based biomaterial-mediated tumor RT, including radiosensitizers and radiopharmaceuticals, with the aim of introducing emerging radiosensitive methods and RT strategies using radiopharmaceuticals. The advantages of peptide-based biomaterials, including controllable synthesis, good biocompatibility, targeting functions, and self-assembly performance, are introduced. These parameters must be considered in the rational design and optimization of peptide-based RT strategies. Peptide-based radiosensitizers are generally divided into three categories: peptides as direct radiosensitizers, peptides as carriers of radiosensitizers, and targeted-peptide-modified radiosensitizers. Peptide-based radiopharmaceuticals are known as peptide-radionuclide conjugates (PRCs) and are categorized according to their peptide targets. Details of PRCs used in clinical studies and US Food and Drug Administration-approved PRCs are also presented. Finally, challenges in the clinical translation of peptide-based biomaterials as RT tools are highlighted.
|
|
Scooped by
?
Today, 12:00 PM
|
This article reviews and provides perspective on the emerging technology of autonomous, ‘self-driving’ laboratories (SDLs) that combine artificial intelligence (AI) and laboratory automation to perform research in chemistry, materials science and biological sciences. Today’s most capable SDLs automate nearly the entire scientific method, from hypothesis generation, experimental design, experiment execution and data analysis, to drawing conclusions and updating hypotheses for subsequent rounds of optimization or discovery. ‘Cloud labs’ offer subscription-based remote-control access to experimental capabilities. Reports of AI-directed experiments executed in cloud labs are appearing in the literature, previewing a democratization of science that intrigues but inspires concern. Indeed, SDLs have potential implications for society far beyond the academy. Inventions emerging from AI-driven science pose a grand challenge, as patent laws across the world recognize only human inventors. If the inventions they generate remain unpatentable, funding for SDLs may be constrained. SDLs raise safety and security concerns. We deem them surmountable with a proactive approach, ultimate human accountability and robust cybersecurity measures. Finally, we estimate the impacts of SDLs on the technical labour force. Our analysis suggests that SDLs may displace some scientific roles but are likely to create many new opportunities.
|
|
Scooped by
?
Today, 11:44 AM
|
An independent evaluation of the spectroscopic properties, cellular performance, merits and pitfalls of the red fluorescent protein mScarlet3-H as compared to mScarlet3 is reported. mScarlet3-H was generated from mScarlet3 by a single M163H mutation. Purified mScarlet3-H is characterized by a molar coefficient of 79,040 M-1cm-1, a fluorescence quantum yield of 17.8%, molecular brightness of 14.1 and a heterogeneous multiexponential decay with an average fluorescence lifetime of 1 ns. Evaluation in living mammalian cells revealed a comparable maturation speed and efficiency of mScarlet3 and mScarlet3-H, but the overall cellular brightness of mScarlet3-H was 5-fold lower than that of mScarlet3. Photobleaching analysis in live cells revealed identical photobleaching kinetics of mScarlet3-H and mScarlet-H. The fluorescence intensity, fluorescence spectra and fluorescence lifetime of mScarlet3-H were found to be strongly pH-dependent between pH 4-8. The fluorescence lifetime increased from 1 ns to 3 ns in lowering the pH from 8 to 4 with a pK of ~6. The much lower lifetime of mScarlet3-H (~1 ns) as compared to mScarlet3 (~4 ns) allows dual fluorescence lifetime unmixing applications in single channel FLIM recordings in compartments with neutral to slightly alkaline pH. Furthermore, the strongly pH-dependent fluorescence lifetime of mScarlet3-H enables fluorescence lifetime-based pH sensing in a pH region between pH 5 to 7. Using this property, a pH of 6.5 was measured in the lumen of the Golgi system in living cells and motile endomembrane vesicles revealed pH values between 5.5 and 6. Also autophagy of the cytoplasm can be visualized by the pH-dependent fluorescence lifetime with mScarlet3-H accumulation in lysosomes. Potential useful applications and pitfalls regarding the special properties of mScarlet3-H are discussed.
|
|
Scooped by
?
Today, 1:44 AM
|
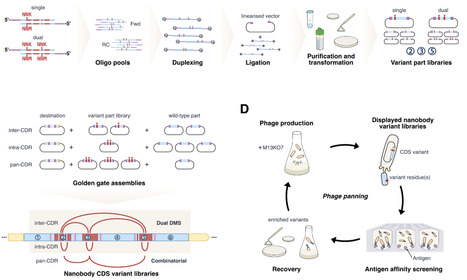
Control over mutational library diversity is an essential consideration when engineering proteins, but is often fraught with trade-offs between diversity, specificity, and affordability. Contemporary library assembly approaches often incorporate oligonucleotide pool synthesis to achieve affordable, precise mutagenesis; however, these oligos are often reliant on complex designs to facilitate downstream PCR and/or restriction digests. Direct hybridization of oligo pools is an overlooked strategy to simplify mutagenesis, especially when paired with a type IIS restriction cloning approach. We validate this approach by designing, hybridising, and deep sequencing single and dual substitution CDR region parts derived from nanobody GA10. Assembly of these parts into a full-length nanobody CDS facilitated the phage display of variant libraries for affinity maturation against its cyclic peptide target. Variants identified through enrichment analysis were expressed in isolation and yielded improved affinities by more than 100-fold. Recent advances in machine learning have successfully inferred improved variants outside of screened library space, but require controlled, multi-mutant libraries. The library assembly approach outlined in this research is well-suited for such approaches.
|
|
Scooped by
?
Today, 1:16 AM
|
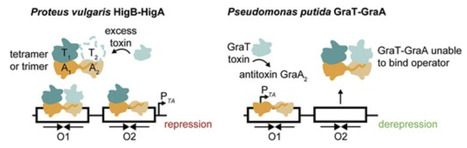
Bacterial toxin–antitoxin (TA) pairs transcriptionally autoregulate their expression via a repression/derepression mechanism in response to changing environmental conditions. The structural diversity of TA systems influences the mechanisms of transcriptional regulation. Here, we define the molecular mechanism for the plasmid-encoded HigB–HigA TA pair originally identified in a post-operative infection with antibiotic-resistant Proteus vulgaris. We determine DNA binding and promoter activity by the HigB–HigA complex supported by structural biology and molecular dynamics simulations of an elusive DNA operator–TA repressor complex. To define the optimal oligomeric TA repressor–DNA operator complex required for derepression, we engineered a dedicated trimeric HigB–HigA2 complex that represses transcription more than 26-fold as compared to the tetrameric HigB2–HigA2. These results expand the known diversity of how the HigB–HigA TA family is autoregulated.
|
|
Scooped by
?
Today, 12:53 AM
|
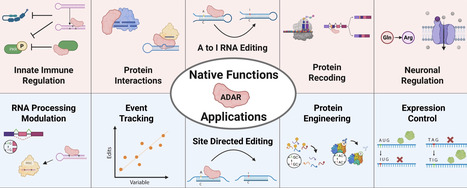
RNA processing is essential for proper cellular function, contributing to protein and cell state diversity, and is often dysregulated in diseased states. A key subset of RNA regulators is the double-stranded RNA-specific adenosine deaminase (ADAR) protein family, which hydrolytically deaminates double-stranded RNA, causing an adenosine-to-inosine edit (A-to-I). Active ubiquitously throughout the body, this pleiotropic protein family plays critical roles in embryonic patterning, neurological function, and immune regulation. Their aberrant activity has in turn been implicated in a spectrum of disorders, including cancer, metabolic diseases, and autoimmune conditions. By instead purposefully modulating their activity, ADARs have been leveraged to create a versatile toolset for transcriptome engineering. This includes enabling programmable RNA editing, controlled RNA splicing, reversibly modulating protein interactions, and altering cellular inflammation. Here, we review the pleiotropic functions and versatile applications of ADARs, as well as outline areas for growth and potential new avenues in both therapeutics and research.
|
|
Scooped by
?
Today, 12:49 AM
|
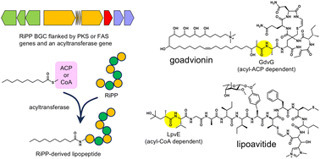
Ribosomally synthesized and post-translationally modified peptides (RiPPs) are a diverse superfamily of natural products unified by a common biosynthetic logic: The peptide backbone is genetically encoded, and the translated precursor peptide undergoes a series of post-translational modifications catalyzed by maturase enzymes to produce the final bioactive compound. Despite their structural complexity, RiPPs are encoded by relatively small biosynthesis gene clusters. RiPP maturase enzymes are diverse and often promiscuous, offering significant biotechnological potential. However, their lack of conserved features makes genome-based discovery of novel RiPPs challenging. Recent advances in biosynthetic understanding and genome mining techniques have led to the identification of numerous uncharacterized RiPP biosynthetic gene clusters, often flanked by genes encoding non-RiPP moieties, in microbial genomes. Leveraging this information, a new class of natural products, hybrids of RiPPs and non-RiPP elements, has recently been discovered. Among them, RiPPs bearing fatty acyl groups, referred to as RiPP-derived lipopeptides, represent a newly emerging class of lipopeptide natural products with significant antimicrobial activity. bgc
|
|
|
Scooped by
?
Today, 4:55 PM
|
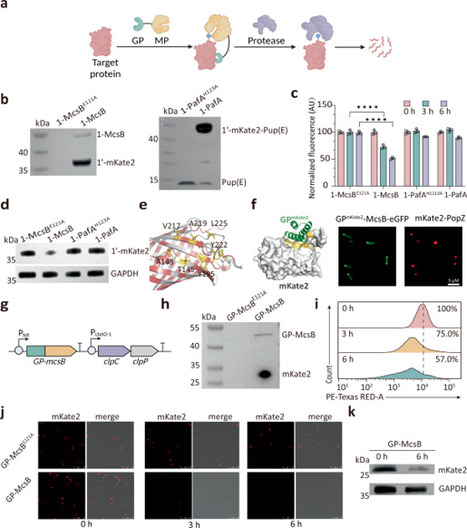
Targeted protein degradation is a powerful tool for biological research, cell therapy, and synthetic biology. However, conventional methods often depend on pre-fused degrons or chemical degraders, limiting their wider applications. Here we develop a guided protein labeling and degradation system (GPlad) in Escherichia coli, using de novo designed guide proteins and arginine kinase (McsB) for precise degradation of various proteins, including fluorescent proteins, metabolic enzymes, and human proteins. We expand GPlad into versatile tools such as antiGPlad, OptoGPlad, and GPTAC, enabling reversible inhibition, optogenetic regulation, and biological chimerization. The combination of GPlad and antiGPlad allows for programmable circuit construction, including ON/OFF switches, signal amplifiers, and oscillators. OptoGPlad-mediated degradation of MutH accelerates E. coli evolution under protocatechuic acid stress, reducing the required generations from 220 to 100. GPTAC-mediated degradation of AroE enhanced the titer of 3-dehydroshikimic acid to 92.6 g/L, a 23.8% improvement over the conventional CRISPR interference method. We provide a tunable, plug-and-play strategy for straightforward protein degradation without the need for pre-fusion, with substantial implications for synthetic biology and metabolic engineering. Targeted protein degradation in bacteria typically requires fusion with tags or chemical degraders. Here, authors developed GPlad, a tunable system using designed guide proteins and arginine kinase to degrade diverse proteins in E. coli without the need for exogenous degraders or protein fusions.
|
|
Scooped by
?
Today, 4:34 PM
|
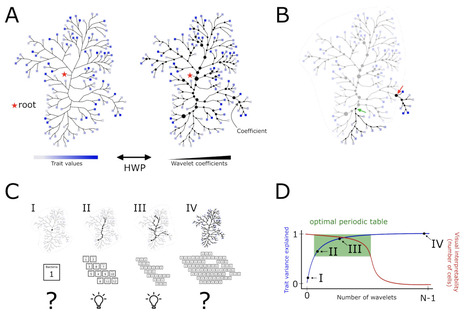
Bacterial diversity can be overwhelming. There is an ever-expanding number of bacterial taxa being discovered, but many of these taxa remain uncharacterized with unknown traits and environmental preferences. This diversity makes it challenging to interpret ecological patterns in microbiomes and understand why individual taxa, or assemblages, may vary across space and time. While we can use information from the rapidly growing databases of bacterial genomes to infer traits, we still need an approach to organize what we know, or think we know, about bacterial taxa to match taxonomic and phylogenetic information to trait inferences. Inspired by the periodic table of the elements, we have constructed a 'periodic table' of bacterial taxa to organize and visualize monophyletic groups of bacteria based on the distributions of key traits predicted from genomic data. By analyzing 50,745 genomes across 31 bacterial phyla, we used the Haar-like wavelet transformation, a model-free transformation of trait data, to identify clades of bacteria which are nearly uniform with respect to six selected traits - oxygen tolerance, autotrophy, chlorophototrophy, maximum potential growth rate, GC content and genome size. The identified functionally uniform clades of bacteria are presented in a concise 'periodic table'-like format to facilitate identification and exploration of bacterial lineages in trait space. While our approach could be improved and expanded in the future, we demonstrate its utility for integrating phylogenetic information with genome-derived trait values to improve our understanding of the bacterial diversity found in environmental and host-associated microbiomes.
|
|
Scooped by
?
Today, 3:23 PM
|
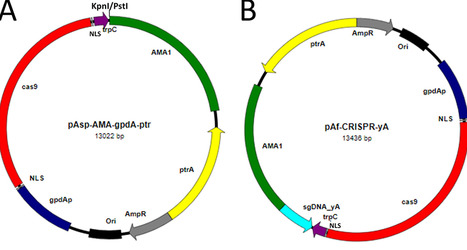
For Aspergillus flavus, a pathogen of considerable economic and health concern, successful gene knockout work for more than a decade has relied nearly exclusively on using nonhomologous end-joining pathway (NHEJ)-deficient recipients via forced double-crossover recombination of homologous sequences. In this study, a simple CRISPR/Cas9 genome editing system that gave extremely high (>95%) gene-targeting frequencies in A. flavus was developed. It contained a shortened Aspergillus nidulans AMA1 autonomously replicating sequence that maintained good transformation frequencies and Aspergillus oryzae ptrA as the selection marker for pyrithiamine resistance. Expression of the codon-optimized cas9 gene was driven by the A. nidulans gpdA promoter and trpC terminator. Expression of single guide RNA (sgRNA) cassettes was controlled by the A. flavus U6 promoter and terminator. The high transformation and gene-targeting frequencies of this system made generation of A. flavus gene knockouts with or without phenotypic changes effortless. Additionally, multiple-gene knockouts of A. flavus conidial pigment genes (olgA/copT/wA or olgA/yA/wA) were quickly generated by a sequential approach. Cotransforming sgRNA vectors targeting A. flavus kojA, yA, and wA gave 52%, 40%, and 8% of single-, double-, and triple-gene knockouts, respectively. The system was readily applicable to other section Flavi aspergilli (A. parasiticus, A. oryzae, A. sojae, A. nomius, A. bombycis, and A. pseudotamarii) with comparable transformation and gene-targeting efficiencies. Moreover, it gave satisfactory gene-targeting efficiencies (>90%) in A. nidulans (section Nidulantes), A. fumigatus (section Fumigati), A. terreus (section Terrei), and A. niger (section Nigri). It likely will have a broad application in aspergilli.
|
|
Scooped by
?
Today, 12:50 PM
|
Unlike tandem repeats in eukaryotes, ribosomal RNA (rRNA) operons across prokaryotic genomes are widely distributed. Here, I examined the distribution of 16S ribosomal RNA gene copies using all entries from the Ribosomal RNA Operon Copy Number Database with a copy number of 2 or greater, using a metric-normalized range-that allows for comparisons between copy number. Normalized range varied across rRNA copy number, with the greatest distribution of rRNA gene copies in prokaryotic accessions with a copy number of 5. There was a significant phylogenetic signal for both bacteria and archaea (p<0.05). Archaea had higher normalized range than bacteria, even when comparing bacteria with comparable copy number (n=2-5) (p<0.05). Normalized range varied across bacterial phyla (p<0.05), with Cyanobacteria having the largest normalized range and Chlamydiae the smallest. Furthermore, normalized range was predicted by copy number, tRNA number, and pseudo-gene number, after correcting for phylum. When restricted to low bacterial copy numbers (n=2-5), normalized range was predicted by genome size, GC content, tRNA number, coding gene number, and pseudo-gene number. For archaea, normalized range was predicted by genome size, GC content, and tRNA number. Comparisons of bacteria and archaea suggest different mechanisms for rRNA copy distribution and maintenance across genomes.
|
|
Scooped by
?
Today, 12:40 PM
|
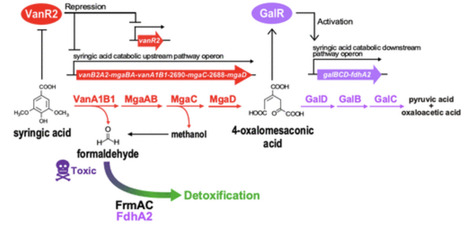
Syringic acid (SA) is a key intermediate in the bacterial catabolism of syringyl lignin-derived aromatic compounds. However, bacterial SA catabolism remains largely unknown. Here, we investigated the SA catabolic system in Pseudomonas sp. NGC7, which catabolizes various lignin-derived aromatic monomers. Pathway analysis and gene disruption studies revealed that SA undergoes O demethylation by VanA1B1 to 3-O-methylgallic acid, which is subsequently catabolized through a linear pathway, involving MgaAB, MgaC, MgaD, GalD, GalB, and GalC into the TCA cycle, unlike the branching pathway found in sphingomonad strains. vanA1B1, mgaAB, mgaC, and mgaD constitute an operon whose transcription is regulated by the GntR-type transcriptional repressor VanR2. In contrast, galBCD constitutes an operon with the formaldehyde dehydrogenase gene fdhA2, and this operon is regulated by the LysR-type transcriptional activator GalR. FdhA2 is involved in the detoxification of formaldehyde, a byproduct of SA catabolism. These findings contribute to the valorization of syringyl lignin-containing biomass.
|
|
Scooped by
?
Today, 12:11 PM
|
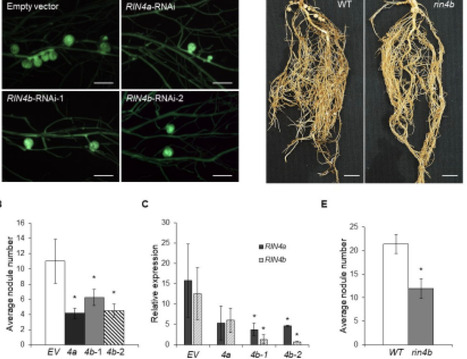
The legume-rhizobium symbiosis represents a unique and beneficial interaction between legumes and nitrogen-fixing soil bacteria, called rhizobia. The initiation and development of this symbiosis is complex and begins with recognition of key molecular signals, produced by the plant and its symbiont, which determine symbiotic compatibility. Current data suggest that the invading symbiont initially triggers plant immune responses that are subsequently suppressed. Hence, there is growing evidence that features of plant immunity may be relevant to symbiotic establishment. RIN4 is a key immune regulator in plants, regulating basal immunity and it is also targeted by pathogen effector proteins that either confer susceptibility or resistance, depending on the presence of the appropriate resistance protein. Surprisingly, we found that RIN4 was rapidly phosphorylated upon rhizobial inoculation of soybean root hairs. RNAi silencing and mutant studies indicate that RIN4 expression is essential for effective nodulation of soybean. RIN4 phosphorylation occurs within a fifteen amino acid motif, which is highly conserved within the Fabales (legumes) and Rosales orders, which comprise species capable of nitrogen-fixing endosymbiosis with rhizobia. RIN4 proteins mutated in this conserved phosphorylation site failed to support efficient soybean nodulation. Phosphorylation of this site is mediated by the symbiotic receptor-like kinase, SymRK, a well-studied member of the symbiotic signaling pathway. The data implicate RIN4 phosphorylation as a key mediator of rhizobial compatibility, interconnecting symbiotic and immune signaling pathways.
|
|
Scooped by
?
Today, 12:02 PM
|
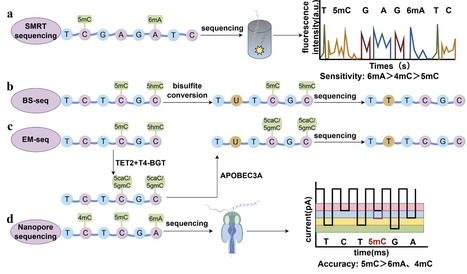
Uncovering the mechanisms regulating the pathogenicity of bacterial pathogens can help improve diagnostic capabilities and aid the development of new drugs, both of which are crucial for reducing the burden caused by bacterial infections. In recent years, with advancements in third-generation sequencing technologies, increasing evidence has shown that DNA methylation plays a pivotal role in the pathogenicity of bacterial pathogens. We believe that the key DNA methyltransferases involved in pathogenicity represent promising targets for antimicrobial therapies and that the DNA methylation sites involved in bacterial pathogenicity are important biomarkers for diagnosing bacterial infections. In this review, we summarize the following topics: (i) methods for DNA methylation sequencing; (ii) the involvement of DNA methylation in antibacterial drug resistance; (iii) the influence of DNA methylation on the expression of bacterial virulence genes; (iv) the impact of DNA methylation on bacterial biofilm formation, adhesion, and motility; and (v) the role of DNA methylation in bacterial adaptation. We hope to provide insights into bacterial pathogenicity from the perspective of bacterial epigenetics.
|
|
Scooped by
?
Today, 11:58 AM
|
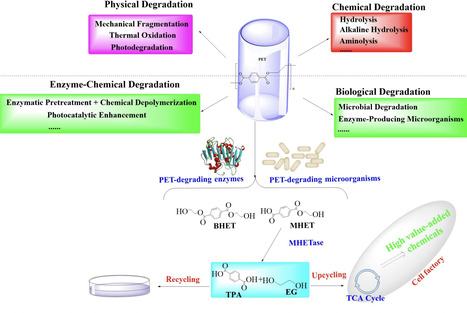
The pervasive accumulation of polyethylene terephthalate (PET) waste has emerged as a critical ecological crisis, which is mainly driven by its recalcitrance to natural degradation and widespread contamination of terrestrial and aquatic ecosystems. In response to this challenge, microbial-mediated PET biodegradation has garnered significant scientific attentions as a sustainable remediation strategy, harnessing the enzymatic cascades of specialized microorganisms to depolymerize PET into bio-assimilable monomers such as terephthalic acid (TPA) and ethylene glycol (EG). In this review, we summarize the extracellular process of PET biodegradation, including microbial attachment, colonization, and direct depolymerization, as well as the metabolic pathways of PET monomers. Strategies for developing PET-degrading chassis cells are also discussed, such as cell surface display, metabolic pathway optimization, and rational design of enzyme-PET interfaces. Microbial-enzyme consortia and molecular engineering of photosynthetic microorganisms also contribute to PET degradation. Although significant progress has been made, challenges remain in enzyme stability, metabolic bottlenecks, industrial scalability, and environmental adaptation. Overall, microbial and enzymatic strategies show great potentials in addressing PET pollution, and future interdisciplinary efforts are needed to overcome these challenges and achieve a sustainable circular plastic economy.
|
|
Scooped by
?
Today, 1:49 AM
|
Human lactoferrin (HLF), a pivotal iron-binding glycoprotein belonging to the transferrin family, is the primary immune protein in breast milk. Currently, recombinant HLF expression in microorganisms has attracted widespread attention. Through genome integration and copy number optimization, this study established a Komagataella phaffii cell factory that achieved an HLF titer of 137.6 mg/L. However, HLF accumulated extensively within the cells. Subsequently, several endogenous signal peptides were screened and hybridized with mating factor α, resulting in a 1.56-fold increase in extracellular HLF titers compared to the control. Additionally, protein kinase II overexpression facilitated vesicle trafficking, further increasing extracellular HLF to a titer of 304.6 mg/L in shake flasks and 1.7 g/L in a 3-L bioreactor. Notably, the intracellular/extracellular HLF concentration ratio decreased from 1.69:1 to 1.01:1. Further enhancements were made through ion optimization and an exponential methanol-feeding strategy, resulting in an extracellular HLF titer of 3.1 g/L. To the best of our knowledge, this is the first study to report considerably improved extracellular HLF production through optimization of the transport and secretion pathways and provide insights into the expression of other secretary human proteins in K. phaffii.
|
|
Scooped by
?
Today, 1:41 AM
|
Recent research has indicated the presence of highly protein occupied, transcriptionally silent regions of bacterial genomes which show functional parallels to eukaryotic heterochromatin. We utilized an integrative approach to track chromatin structure and transcription in Escherichia coli K-12 across a wide range of nutrient conditions. In the process, we identified multiple loci which act similarly to facultative heterochromatin in eukaryotes, normally silenced but permitting expression of genes under specific conditions. We also found a strong enrichment of small regulatory RNAs (sRNAs) among the set of differentially expressed transcripts during nutrient stress. Using a newly developed bioinformatic pipeline, the transcription factors (TFs) regulating sRNA expression were bioinformatically predicted, with experimental follow-up revealing novel relationships for 45 sRNA–TF candidates. Direct regulation of sRNA expression was confirmed by mutational analysis for five sRNAs of metabolic interest: IsrB (also known as AzuCR), CsrB and CsrC, GcvB, and GadY. Our integrative analysis thus reveals additional layers of complexity in the nutrient stress response in E. coli and provides a framework for revealing similar poorly understood regulatory logic in other organisms.
|
|
Scooped by
?
Today, 1:14 AM
|
CRISPR–Cas12a enzymes are versatile RNA-guided genome-editing tools with applications encompassing viral diagnosis, agriculture, and human therapeutics. However, their dependence on a 5′-TTTV-3′ protospacer adjacent motif (PAM) next to DNA target sequences restricts Cas12a’s gene targeting capability to only ∼1% of a typical genome. To mitigate this constraint, we used a bacterial-based directed evolution assay combined with rational engineering to identify variants of Lachnospiraceae bacterium Cas12a with expanded PAM recognition. The resulting Cas12a variants use a range of noncanonical PAMs while retaining recognition of the canonical 5′-TTTV-3′ PAM. In particular, biochemical and cell-based assays show that the variant Flex-Cas12a utilizes 5′-NYHV-3′ PAMs that expand DNA recognition sites to ∼25% of the human genome. With enhanced targeting versatility, Flex-Cas12a unlocks access to previously inaccessible genomic loci, providing new opportunities for both therapeutic and agricultural genome engineering.
|
|
Scooped by
?
Today, 12:50 AM
|
In this Journal Club, Jian Shu recalls a 2006 publication by Takahashi and Yamanaka as well as a 2021 paper introducing AlphaFold to discuss the fascinating potential of cellular reprogramming in the age of artificial intelligence.
|
















omv, 2st, idea