 Your new post is loading...

|
Scooped by
?
Today, 12:53 AM
|
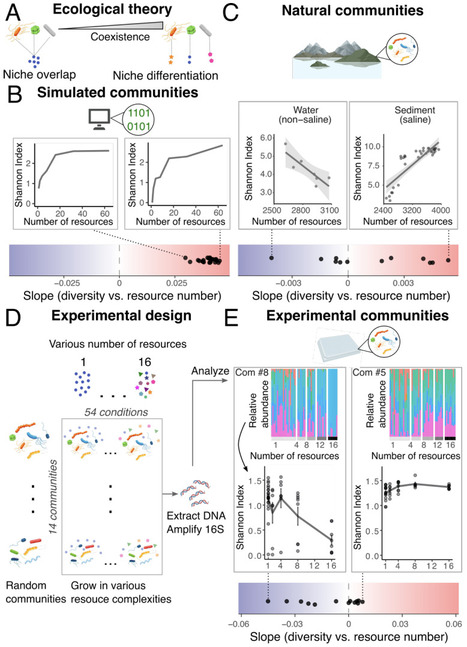
The origin of biodiversity is arguably the most fundamental question in ecology - especially how countless microbial species coexist within a single community. A prevailing hypothesis holds that microbial species coexist by specializing on different resources, thereby reducing competition. Accordingly, increasing resource diversity is expected to promote species coexistence and boost biodiversity. Surprisingly, we observe the opposite: microbial biodiversity often declines as resource diversity increases. This unexpected result emerges from a widespread physiological response across diverse microbial taxa, in which more complex environments trigger higher overall resource uptake. This intensifies competition and drives biodiversity loss. Updating a central ecological model to include this physiological trait accurately reproduces the observed decline. Our findings demonstrate that physiological changes at the individual level can overturn classical ecological principles and scale up to restructure entire ecosystems.
|
|
Scooped by
?
Today, 12:17 AM
|
Dominance of Bacillus species on conventional agar media often hampers the isolation of taxonomically diverse soil bacteria (i.e., 'Bacillus dominance'). To overcome this Bacillus dominance, we developed a novel agar medium containing leaf mold extract (LME) and assessed its utility for isolating microorganisms from soil samples. For comparison, conventional yeast malt extract agar (YME agar) was used as a control. 16S rRNA gene sequencing revealed that colonies cultured on YME agar predominantly consisted of Bacillus-related bacteria, whereas LME agar enabled the growth of a wider variety of taxa, particularly actinomycetes such as Streptomyces. Notably, LME agar supported a higher diversity of soil bacterial isolates even from samples with a low abundance of Bacillus-related species. Among the 138 isolates recovered from LME agar, some either exhibited very low 16S rRNA gene sequence identity to known species in public databases, or failed to yield 16S rRNA amplicons with conventional universal primers, suggesting the presence of previously uncharacterized or novel taxa. Furthermore, one unidentified isolate failed to grow on standard nutrient media (YME, BHI, LB10, or TSB), but proliferated in liquid leaf mold extract, indicating a specific nutrient dependency. These findings demonstrate that LME agar can facilitate the isolation of a broader spectrum of soil microorganisms (including rare and previously uncultured species) not readily recoverable on common basal media. Incorporation of ecosystem-derived substrates into culture media may greatly enhance the discovery and study of novel microbial diversity from environmental samples.
|
|
Scooped by
?
Today, 12:04 AM
|
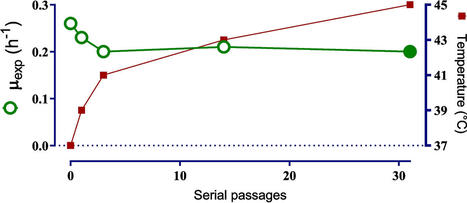
The heat shock response is a cellular protection mechanism against sudden temperature upshifts extensively studied in Escherichia coli. However, the effects of thermal evolution on this response remain largely unknown. In this study, we investigated the early and late physiological and transcriptional responses to temperature upshift in a thermotolerant strain under continuous culture conditions. Adaptive laboratory evolution ALE was performed on a metabolically engineered E. coli strain (JU15), designed for d-lactic acid production, to enable cellular growth and fermentation of glucose at 45 °C in batch cultures. The resulting homofermentative strain, ECL45, successfully adapted to 45 °C in a glucose-mineral medium at pH 7 under non-aerated conditions. The thermal-adapted ECL45 retained the parental strain’s high volumetric productivity and product/substrate yield. Genomic sequencing of ECL45 revealed eight mutations, including one in a non-coding region and six within the coding regions of genes associated with metabolic, transport, and regulatory functions. Transcriptomic analysis comparing the evolved strain with its parental counterpart under early and late temperature upshifts indicated that the adaptation involved a controlled stringent response. This mechanism likely contributes to the strain’s ability to maintain growth capacity at high temperatures.
|
|
Scooped by
?
May 13, 11:42 PM
|
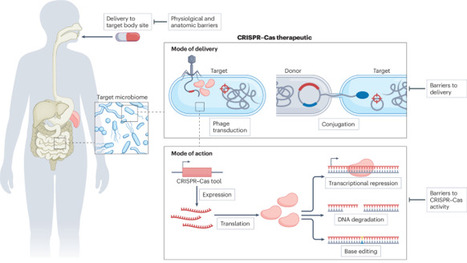
Technologies derived from the CRISPR–Cas immune system of prokaryotes have revolutionized our ability to cleave and modify target nucleic acid sequences. In addition to the use of CRISPR–Cas tools for the editing of human genes, they can also be designed to target pathogenic and commensal bacteria that colonize the body, offering new pathways for the treatment of infections and microbiome modulation. In this Review, we explore how the CRISPR–Cas toolbox can be engineered to kill or modify specific bacteria. We discuss DNA-targeting and RNA-targeting strategies, outlining how these can be applied to disarm bacteria by removing, modifying or silencing specific genes. Furthermore, we examine the delivery of CRISPR–Cas tools by bacteriophages and through conjugation and explore intracellular barriers to CRISPR–Cas tool maintenance and expression. Finally, we highlight therapeutic opportunities in the treatment of infectious diseases and for the modification of the microbiome, outlining progress and challenges in translating these approaches into clinical applications. CRISPR–Cas tools can be designed to kill or modify specific bacteria. This Review explores the engineering of CRISPR–Cas tools and corresponding delivery strategies for the treatment of bacterial infections and modification of the microbiome.
|
|
Scooped by
?
May 13, 11:33 PM
|
All methanogens that can fix nitrogen use molybdenum (Mo) nitrogenase. Some methanogens, including Methanosarcina acetivorans, also contain alternative vanadium- and iron-nitrogenases, encoded by the vnf and anf operons, respectively. These nitrogenases are produced when there is insufficient Mo to support Mo-nitrogenase activity. The factors that control the expression of the alternative nitrogenases in response to Mo availability are unknown in methanogens. Here we show that ModE is the regulator that represses transcription of the vnf and anf operons in M. acetivorans when cells are grown with Mo. CRISPRi repression of modE results in a significant increase in the transcription of the vnf and anf operons as well as the detection of Fe-nitrogenase during nitrogen fixation in the presence of Mo. Gel shift assays with recombinant ModE demonstrated that ModE binds a specific sequence motif upstream of the vnf and anf operons, as well as other genes and operons related to nitrogen fixation and Mo transport. However, purified ModE does not contain Mo, and the addition of Mo does not alter the affinity of ModE for DNA, indicating M. acetivorans ModE may not directly bind Mo. This study shows that ModE is the primary Mo-responsive regulator of alternative nitrogenase expression in M. acetivorans, but other factor(s) are likely involved in directly sensing Mo.
|
|
Scooped by
?
May 13, 11:19 PM
|
Bacterial phenotypic plasticity enables rapid adaptation to fluctuating environments. Filamentation, a shape-shifting response commonly observed under stress, has often been viewed as a byproduct of cellular damage. However, filamentation might also confer survival advantages by influencing toxin accumulation dynamics. In this study, we examined the adaptive value of filamentation in Escherichia coli using a genetically controlled, SOS-independent induction system to compare isogenic, yet phenotypically distinct cells. By integrating mathematical modeling, single-cell microfluidics, and time-resolved flow cytometry, we evaluate bacterial survival under heavy metal and β-lactam antibiotic stress. Our results show that filamentation can improve survival by decreasing the surface area-to-volume ratio, which slows intracellular toxin accumulation and extends the time available for stress response activation or for external toxin levels to dissipate. These findings suggest that filamentation serves as an effective morphological strategy to transiently withstand environmental toxicity, reinforcing its broader role in bacterial stress adaptation.
|
|
Scooped by
?
May 13, 11:11 PM
|
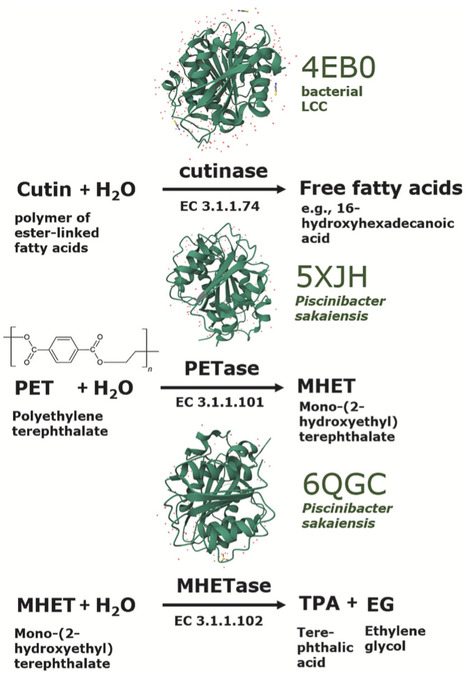
Biological degradation of plastics by microbial enzymes offers a sustainable alternative to traditional waste management methods that often pollute the environment. This review explores ecologically-informed bioprospecting for microorganisms possessing enzymes suitable for biological plastic waste treatment. Natural habitats enriched in plastic-like polymers, such as insect-derived polyesters, epicuticular microbial biofilms in the phyllosphere of plants in extreme environments, or aquatic ecosystems, are highlighted as promising reservoirs for bioprospecting. Anthropogenic habitats, including plastic-polluted soils and the plastisphere, have yielded potent enzymes such as PETases and cutinases, which are being exploited in biotechnology. However, bioprospecting in plastispheres and artificial environments frequently leads to the isolation of environmental opportunistic microorganisms, such as Pseudomonas aeruginosa, Aspergillus fumigatus, Parengyodontium album, or species of Fusarium, which are capable of becoming human and/or plant pathogens. These cases necessitate stringent biosecurity measures, including accurate molecular identification, ecological assessment, and containment protocols. Beyond advancing bioprospecting approaches toward a broader scope of relevant habitats, this review underscores the educational value of such screenings, specifically, in understudied natural habitats, emphasizing its potential to uncover novel enzymes and microorganisms and engage the next generation of researchers in interdisciplinary study integrating environmental microbiology, molecular biology, enzymology, polymer chemistry, and bioinformatics. Finally, we offer guidelines for microbial bioprospecting in various laboratory settings, ranging from standard environmental microbiology facilities to high-biosecurity facilities, thereby maximizing the diversity of scientists who may contribute to addressing urgent environmental challenges associated with plastic waste.
|
|
Scooped by
?
May 13, 10:01 PM
|
De novo design of anti-freeze peptides (AFPTs) represents a formidable challenge due to the unclarified active structure of AFPTs. Here, we describe a “Site to Distance” principle for de novo design of AFPTs, in terms of understanding their structure–activity relationships. The first step is to point E, identified as the most potent ice-binding site (IBS) possessing at least 4-fold binding energy than natural IBSs, into the candidate backbones. The second step, based on the IBS (E), is to judiciously adjust the distances of sites to match the favorable number of the ice crystal lattice to achieve the strongest ice-binding, relying on a newly established low-temperature AFPT structure prediction platform. The resultant AFPTs show a substantial reduction in single ice crystal growth rates, much superior to >100 natural or designed AFPTs, including all that have been reported. Cryopreservation of therapeutic cells further confirms the accuracy of this design principle.
|
|
Scooped by
?
May 13, 9:32 PM
|
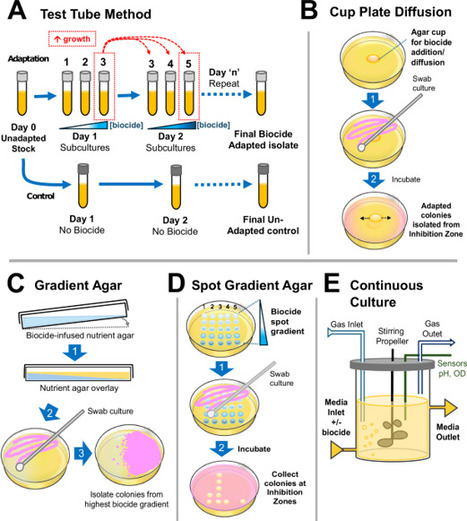
Decreased bacterial susceptibility to biocides raises concerns due to their influences on antibiotic resistance. The lack of standardized breakpoints, established methods, and consistent terminology complicates this research. This review summarizes techniques for studying biocide resistance mechanisms, susceptibility testing, and in-vitro adaptation methods, highlighting their benefits and limitations. Here, the challenges in studying biocide susceptibility and the need for standardized approaches in biocide research are emphasized for commonly studied biocide classes.
|
|
Scooped by
?
May 13, 9:15 PM
|
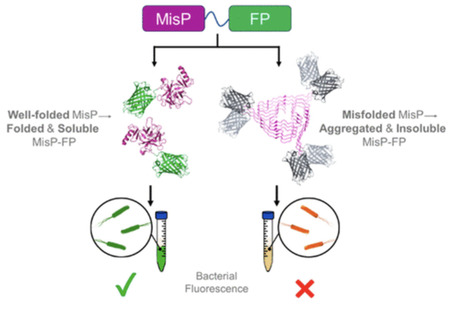
Protein misfolding and aggregation are central features of a wide range of diseases, including neurodegenerative disorders, systemic amyloidoses, and cancer. The identification of compounds that can modulate protein folding and aggregation is a key step toward developing effective therapies. High-throughput screening methods are essential for efficiently identifying such compounds. In this study, we optimized a previously developed high-throughput genetic screen for monitoring protein misfolding and aggregation in bacteria. This system is based on monitoring the fluorescence of Escherichia coli cells expressing fusions of human misfolding-prone and disease-related proteins (MisPs) with the green fluorescent protein. We systematically tested a variety of experimental conditions, such as overexpression conditions and MisP-GFP fusion formats, to identify key parameters that affect the sensitivity and dynamic range of the assay. Using misfolding-prone, cancer-associated variants of human p53 as a model system, we found that strong overexpression conditions, such as high copy number vectors, strong promoters, high inducer concentrations, and high overexpression temperatures, can yield optimal assay performance. These optimized assay conditions were also validated with additional MisPs, such as the Alzheimer’s disease-associated amyloid-β peptide and variants of superoxide dismutase 1 associated with amyotrophic lateral sclerosis. At the same time, we observed that certain conditions, such as inducer concentrations and overexpression temperature, may need to be precisely fine-tuned for each new MisP target to yield optimal assay performance. Our findings provide a framework for standardizing MisP-GFP screening assays, facilitating their broad application in the discovery of therapeutic agents targeting protein misfolding and aggregation.
|
|
Scooped by
?
May 13, 5:53 PM
|
The common fungal pathogen, Candida albicans, relies on the pore-forming toxin candidalysin to damage host cells. Cells resist other pore-forming toxins by Ca2+ dependent microvesicle shedding and annexins (cholesterol-dependent cytolysins, CDCs), or annexins and patch repair (aerolysin). However, it is unclear which Ca2+ dependent repair mechanism(s) resists candidalysin. Here, we determined the involvement of different Ca2+ dependent repair mechanisms to candidalysin and compared responses to CDCs and aerolysin using flow cytometry and high-resolution microscopy. We report that candidalysin triggered Ca2+-dependent repair, but patch repair and ceramide failed to provide significant protection. MEK-dependent repair and annexins A1, A2 and A6 contributed partially to repairing damage caused by candidalysin. However, annexin translocation after candidalysin challenge was delayed compared to CDCs or aerolysin challenge. Surprisingly, extracellular Cl- improved cell survival after candidalysin challenge, but not after challenge with a CDC or aerolysin. Finally, we found that candidalysin is removed via extracellular vesicle shedding. These findings reveal that Ca2+ dependent microvesicle shedding protects cells from candidalysin and can be engaged by multiple molecular mechanisms during membrane repair.
|
|
Scooped by
?
May 13, 3:48 PM
|
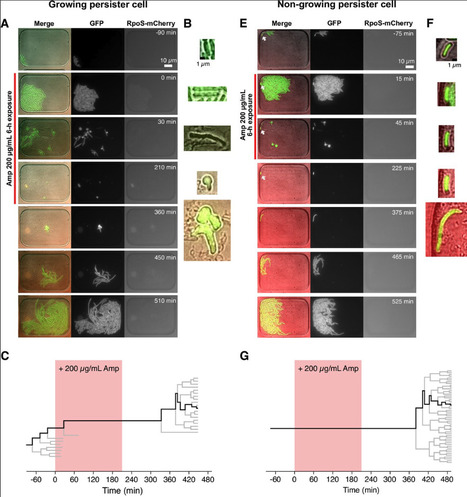
Bacterial persistence is a phenomenon in which a small fraction of isogenic bacterial cells survives a lethal dose of antibiotics. Although the refractoriness of persistent cell populations has classically been attributed to growth-inactive cells generated before drug exposure, evidence is accumulating that actively growing cell fractions can also generate persister cells. However, single-cell characterization of persister cell history remains limited due to the extremely low frequencies of persisters. Here, we visualize the responses of over one million individual cells of wildtype Escherichia coli to lethal doses of antibiotics, sampling cells from different growth phases and culture media into a microfluidic device. We show that when cells sampled from exponentially growing populations were treated with ampicillin or ciprofloxacin, most persisters were growing before antibiotic treatment. Growing persisters exhibited heterogeneous survival dynamics, including continuous growth and fission with L-form-like morphologies, responsive growth arrest, or post-exposure filamentation. Incubating cells under stationary phase conditions increased both the frequency and the probability of survival of non-growing cells to ampicillin. Under ciprofloxacin, however, all persisters identified were growing before the antibiotic treatment, including samples from post-stationary phase culture. These results reveal diverse persister cell dynamics that depend on antibiotic types and pre-exposure history.
|
|
Scooped by
?
May 13, 3:11 PM
|
Accurate and accessible phylogenetic analysis is essential for understanding microbial taxonomy and evolution, which are integral to microbiology, ecology, and drug discovery, yet it remains a challenging task. AutoMLST2 https://automlst2.ziemertlab.com is a web server designed to facilitate automated phylogenetic reconstruction and microbial taxonomy analysis for bacterial and archaeal genomes. It builds on the foundation of AutoMLST, which remains widely used due to its user-friendly interface compared to similar tools. Given its continued popularity and utility, we have enhanced AutoMLST to leverage newer reference databases and computational tools. AutoMLST2 integrates the Genome Taxonomy Database, extends support to archaeal genomes, and improves analytical flexibility. Key improvements include more customizable processing modes, containerization to prevent queue accumulations, and parallel computing for large-scale studies. By incorporating up-to-date databases and workflows, AutoMLST2 continues to provide an accessible and efficient platform for researchers in microbiology, evolutionary ecology, and natural product discovery.
|
|
|
Scooped by
?
Today, 12:47 AM
|
Robust control of DNA replication is fundamental to bacterial proliferation. In Escherichia coli, replication initiation is thought to be regulated by oscillations in DnaA activity, driven by DnaA-chromosome interactions that differ among leading models. However, direct evidence linking these oscillations to replication initiation has been lacking, and existing models fail to explain the observed decoupling of replication initiation from dnaA expression. Here, we establish a direct link between DnaA activity and replication initiation by demonstrating robust oscillations in DnaA activity, which peak precisely at replication initiation across diverse growth conditions and genetic perturbations. Notably, these oscillations persist even when dnaA transcription remains constant, suggesting a regulatory mechanism that modulates DnaA activity independently of its expression. Additionally, we propose an extrusion model in which DNA-binding proteins sense biomass-DNA imbalance and extrude DnaA from the chromosome to trigger replication, overcoming limitations of existing models. Consistent with this model, perturbation of the nucleoid-associated protein H-NS modulates DnaA activity and replication timing, supporting its mechanistic validity.
|
|
Scooped by
?
Today, 12:12 AM
|
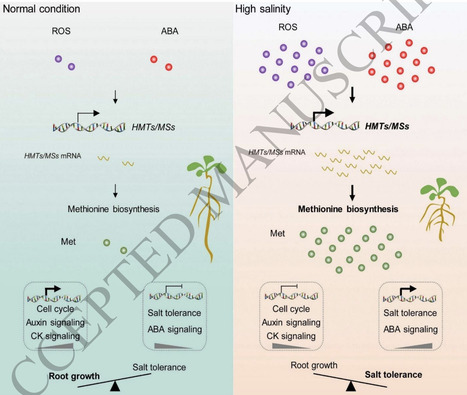
the methionine-mediated balance between salt tolerance and root growth. Under normal conditions, low levels of ROS and ABA signaling maintain low methionine synthesis, promoting root growth through activating cell cycle, auxin, and cytokinin pathways while repressing salt tolerance genes. In contrast, high salinity activates ROS and ABA signaling, upregulates methionine biosynthesis, and elevates methionine levels. This shift enhances salt tolerance by boosting ABA signaling and salt-responsive gene expression, while simultaneously inhibiting root growth by suppressing genes involved in cell division and hormone signaling
|
|
Scooped by
?
May 13, 11:58 PM
|
The gut microbiota is critical for neutralizing dietary toxins. Oxalate is a toxin commonly produced by plants to deter herbivory and is widely consumed in the human diet. Excess levels of systemic or urinary oxalate increase risk of multiple urologic and cardiometabolic diseases. The current study employed multiple amplicon-based and shotgun metagenomic methodologies, alongside comparative phylogenetic analyses, to interrogate evolutionary radiation surrounding microbial oxalate degradation within the human gut microbiome. In conservative genome-based estimates, over 30% of gut microbial species harbored at least one oxalate-handling gene, with the specific pathways used dependent on bacterial phylum. Co-occurrence analyses revealed interactions between specialist genes that can metabolize oxalate or its by-products, but not multi-functional genes that can act in more than one oxalate-related pathway. Specialization was rare at the genome level. Amplicon-based metagenomic sequencing of the oxalate-degrading gene, formyl-CoA transferase (frc), coupled with molecular clock phylogenetic analyses are indicative of rapid evolutionary divergence, constrained by phylum. This was corroborated by paired analyses of non-synonymous to synonymous substitutions (dN/dS ratios), which pointed toward neutral to positive selection. Sequence similarity network analyses of frc sequences suggest extensive horizontal gene transferring has occurred with the frc gene, which may have facilitated rapid divergence. The frc gene was primarily allocated to the Pseudomonodota phylum, particularly the Bradyrhizobium genus, which is a species capable of utilizing oxalate as a sole carbon and energy source. Collectively evidence provides strong support that, for oxalate metabolism, evolutionary selection occurs at the gene level, through horizontal gene transfer, rather than at the species level.
|
|
Scooped by
?
May 13, 11:38 PM
|
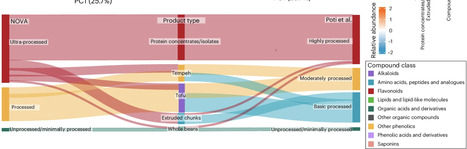
According to existing food processing classification systems, plant-based protein-rich (PBPR) foods are often considered ‘ultra-processed’—and therefore perceived as unhealthy—despite their ability to provide various bioactive compounds beneficial for human health. Here we used a non-targeted metabolomics approach to analyse the impact of processing on the biochemical composition of PBPR foods. Our results show that existing food classification systems may provide questionable categories for PBPR foods without considering their overall biochemical composition, including phytochemicals. An analysis focusing specifically on biochemical compounds of soy-based products manufactured using various technologies showed no clear distinctions between processing groups in the principal component analysis based on the NOVA and Poti classification. However, clear differences were found between soy-based products based on their phytochemical profile. Although food processing classification systems are welcome in their attempt to guide consumers towards healthy choices, they should be improved to more accurately reflect the biochemical composition of PBPR foods. Although plant-based protein-rich (PBPR) foods contain nutritionally bioactive compounds, they are often classified as ultra-processed foods, which most consumers perceive as unhealthy. Using a non-targeted metabolomic approach, this study shows that existing food classification systems do not consider the biochemical composition of PBPR foods, potentially misleading consumers to avoid these products.
|
|
Scooped by
?
May 13, 11:25 PM
|
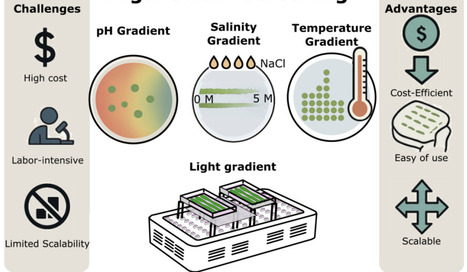
Bioprospecting algae strains with tolerance to extreme conditions such as pH, temperature, salinity, and light is crucial for advancing biotechnology and environmental applications. However, traditional screening methods often involve significant costs and labor, restricting their accessibility and practical use. In this study, we developed and validated low-cost, high-throughput screening techniques, predominantly employing agar plates and liquid culture assays, to effectively differentiate tolerance levels among various algae strains. The methodologies were optimized using the model microalga Chlamydomonas reinhardtii and its closely related species Chlamydomonas incerta and the recently discovered extremophilic Chlamydomonas pacifica. We systematically evaluated the algae for tolerance to extremes by establishing precise gradients of pH (acidic to alkaline conditions), salinity (0 to 5 M NaCl), temperature (34 - 42C), and light intensity (40 to 2977 microE/m2s1). Our results demonstrated that these cost-effective, agar plate-based methods effectively distinguished algae strains exhibiting superior tolerance to extreme environmental conditions. These screening techniques not only provided clear differentiation among the closely related strains but also delivered reproducible outcomes suitable for scaling up to larger bioprospecting efforts. Furthermore, the affordability and simplicity of these methods facilitate their implementation in resource-limited laboratories, thereby broadening participation in algae bioprospecting endeavors. This study highlights the potential of low-cost, accessible screening techniques to significantly enhance the discovery and characterization of algal strains with extreme traits. Ultimately, these methods support the development of robust algae-based resources, driving innovation in diverse industrial processes and environmental solutions.
|
|
Scooped by
?
May 13, 11:15 PM
|
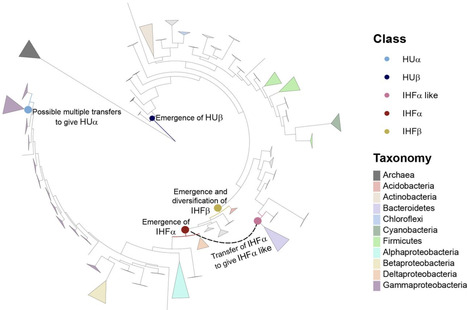
Transcriptional regulation enables bacteria to adjust to its environment. This is driven by transcription factors (TFs), which display DNA site recognition specificity with some flexibility built in. TFs, however, are not considered essential to a minimal cellular life. How did they evolve? It has been hypothesized that TFs evolve by gaining specificity (and other functions) on a background of non-specific chromosome structuring proteins. We used the IHF/HU family of DNA binding proteins, in which IHF binds DNA in a sequence-specific manner, whereas HU binds more indiscriminately, to test this hypothesis. We show that HUβ has been present from the bacterial root, while both IHF subunits emerged much later and diversified in Proteobacteria, with HUα having possibly arisen from transfer events in Gammaproteobacteria. By reconstructing ancestral sequences in-silico on a rooted phylogeny of IHF/HU we show that the common ancestor of this family was probably HU-like and therefore non-specific in binding DNA. IHF evolved from a branch of HU after HU had substantially diverged. Various residues characteristic of IHFα and shown to be involved in specific sequence recognition (at least in E. coli) have likely been co-opted from preexisting residues in HU, while those residues of IHFβ have likely evolved independently, suggesting that each of the IHF subunits has undergone different trajectories to acquire their DNA binding properties.
|
|
Scooped by
?
May 13, 10:16 PM
|
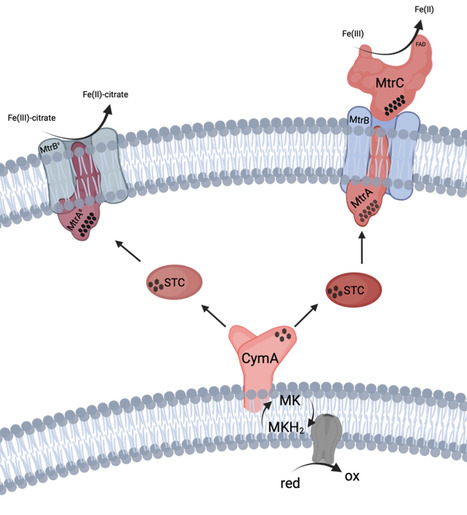
Research in electro-microbiology provides unique opportunities to study and exploit microbial physiology. Several efforts have been made to transplant the extracellular electron transport chain from the native exoelectrogenic model organism Shewanella oneidensis into Escherichia coli. However, systematic comparisons between donor and recipient strain configurations are largely missing. Hence, the proposed minimal protein set, consisting of the c-type cytochromes cytoplasmic membrane protein A (CymA), small tetraheme cytochrome (STC), MtrA, and MtrC, as well as the β-barrel protein MtrB, was heterologously expressed in E. coli in different expansion stages. These stages were compared to corresponding S. oneidensis strains in terms of anthraquinone-2,6-disulfonate (AQDS) and ferric citrate reduction rates. This revealed that transplantation of heterologous extracellular electron transfer (EET) chains is associated with a tremendous decrease in electron transfer rates. As the acquired electron transfer rates were not competitive to S. oneidensis, it was hypothesized that protein localization and maturation might be affected by heterologous expression. Hence, the type II secretion system from S. oneidensis was also transplanted into an E. coli. The latter allowed the secretion of the terminal reductase MtrC onto the cell surface of E. coli for the first time. This was correlated with significantly increased but still insufficient extracellular electron transfer rates. Further experiments suggest that the correct folding of MtrB might be a further bottleneck.
|
|
Scooped by
?
May 13, 9:35 PM
|
Advances in cellular physiology research have created the need to visualize metabolic processes in both time and space. Vibrational microscopy has emerged as a promising method at the forefront of this field. Here, we summarize recent progress and offer perspectives on the application of vibrational microscopy for uncovering metabolic actions in vivo.
|
|
Scooped by
?
May 13, 9:24 PM
|
Oligonucleotide synthesis serves as a cornerstone of modern life sciences, enabling groundbreaking advancements across molecular diagnostics, therapeutic development, and transformative technologies including DNA data storage and programmable biological systems. While phosphoramidite-based chemical synthesis remains the industrial standard, its limitations in producing long-sequence constructs, cumulative error rates, and reliance on toxic solvents pose significant challenges for next-generation applications. Emerging enzymatic synthesis approaches offer a paradigm shift by harnessing the inherent precision and environmental sustainability of biological systems. This comprehensive review systematically examines the evolving landscape of oligonucleotide synthesis technologies. We first analyze the mechanistic foundations and persistent limitations of conventional chemical methods, followed by a critical evaluation of enzymatic strategies with particular emphasis on terminal deoxynucleotidyl transferase (TdT)-mediated template-independent polymerization. The work provides detailed insights into enzymatic reaction engineering, including substrate specificity profiling of nucleotide analogs and innovative solid-phase synthesis platforms enabling iterative nucleotide addition. Furthermore, we discuss emerging high-throughput synthesis architectures and commercial translation efforts. In summary, this review comprehensively encapsulates the advancements and commercialization status of enzymatic synthesis technologies, offering valuable guidance that can expedite the innovative development of enzymatic oligonucleotide manufacturing platforms.
|
|
Scooped by
?
May 13, 9:08 PM
|
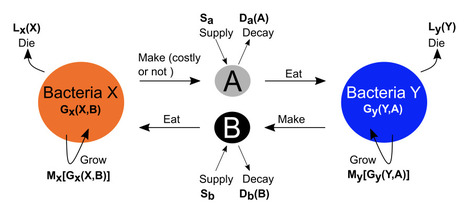
Bacterial cooperation involves the exchange of metabolites, which can range from costless byproducts of metabolism to intentionally produced and costly molecules. These interactions occur across spatial scales, from direct cell contact to the diffusion of metabolites in the environment. Due in part to this variety of interaction modes, the impacts of mutualism on bacterial community dynamics remain unclear. Using generalized models, we derive conditions for the onset of different dynamical behaviors in communities of bacterial cooperators across these scenarios. These include exchanges of low-cost metabolites, costly cross-feeding, and cases where bacteria produce either most or only a small fraction of available metabolites. Stability depends strongly on metabolic production costs and the balance between metabolite uptake and production. We further show that perturbations to bacteria have larger impacts than to metabolites. Finally, we demonstrate that spatial metabolite diffusion drives pattern formation, emphasizing the links between local stability and spatial structures in bacterial cooperation.
|
|
Scooped by
?
May 13, 5:45 PM
|
The genomes of flowering plants consist largely of transposable elements (TEs), some of which modulate gene regulation and function. However, the repetitive nature of TEs and difficulty of mapping individual TEs by short-read sequencing have hindered our understanding of their regulatory potential. Here we show that long-read chromatin fibre sequencing (Fiber-seq) comprehensively identifies accessible chromatin regions (ACRs) and CpG methylation across the maize genome. We uncover stereotypical ACR patterns at young TEs that degenerate with evolutionary age, resulting in TE enhancers preferentially marked by a novel plant-specific epigenetic feature: simultaneous hyper-CpG methylation and chromatin accessibility. We show that TE ACRs are co-opted as gene promoters and that ACR-containing TEs can facilitate gene amplification. Lastly, we uncover a pervasive epigenetic signature—hypo-5mCpG methylation and diffuse chromatin accessibility—directing TEs to specific loci, including the loci that sparked McClintock’s discovery of TEs. Long-read chromatin assay reveals (1) a novel class of accessible chromatin regions, (2) accessibility within individual LTR retrotransposons and (3) the relationship between diffuse accessibility, gene body methylation and hAT transposon insertion.
|
|
Scooped by
?
May 13, 3:40 PM
|
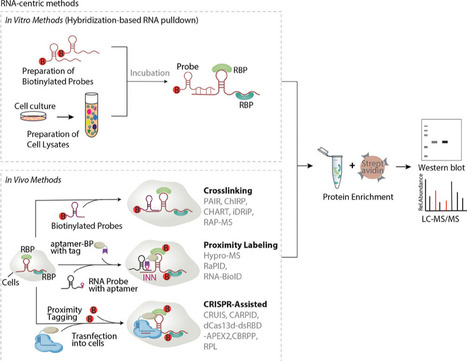
RNA–protein interactions are fundamental to cellular processes such as gene regulation and RNA metabolism. Over the past decade, significant advancements in methodologies have transformed the ability to study these interactions with unprecedented resolution and specificity. This review systematically compares RNA- and protein-centric approaches, highlighting their strengths, limitations, and optimal applications. RNA-centric methods, including hybridization-based pulldowns, proximity labeling, and CRISPR-assisted techniques, enable the identification of proteins interacting with specific RNAs, even low-abundance or transient partners. Protein-centric strategies, such as immunoprecipitation-based CLIP-seq, and emerging proximity-tagging methods, map RNA interactomes of RNA-binding proteins with nucleotide precision. This study evaluates key innovations like LACE-seq and ARTR-seq, which minimize cell input requirements, and HyPro-MS, which bypasses genetic modifications. Guidelines for method selection are provided, emphasizing experimental goals, RNA abundance, interaction dynamics, and technical constraints. Critical challenges are also discussed, including capturing low-affinity interactions, resolving RNA structural complexities, and integrating multi-omics data. This review underscores the importance of method-tailoring to biological contexts, offering a roadmap for researchers to navigate the evolving landscape of RNA–protein interaction studies. By bridging technical advancements with practical recommendations, this study aims to accelerate discoveries in RNA biology, therapeutic development, and precision medicine.
|


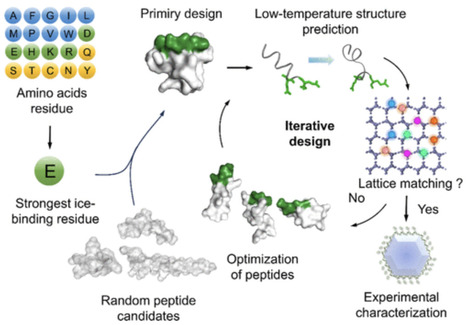


















cryoprotectant, First, “site incorporation” means replacing the residues with E or just adding E into the peptide sequence. After a full investigation of all 20 natural amino acid residues, E is the most potent ice-binding site (IBS), presenting 4-fold strength compared to natural IBS, such as T. Second is “distance tuning”, where a low-temperature conformational computational design platform is established to get precise AFPT structures under the ice nucleation temperatures, ensuring the distance between IBSs matches the favorable number of lattices of the ice plane