Your new post is loading...
Your new post is loading...

|
Scooped by
I2BC Paris-Saclay
October 17, 2024 6:14 AM
|
Introduction to biological crystallography
Following the French MOOC 'Voyage au cœur du vivant avec des rayons X : la cristallographie', available on FUN-MOOC from 2017 to 2022, and the book in French 'Introduction à la cristallographie biologique', we now present an English version. Following the French MOOC 'Voyage au cœur du vivant avec des rayons X : la cristallographie', available on FUN-MOOC from 2017 to 2022, and the book in French 'Introduction à la cristallographie biologique', we now present an English version.
This book is primarily designed for beginners in biological crystallography.
It provides an introduction to the different steps of biological crystallography, from the crystallisation to the three-dimensional structure of a macromolecule. The QR codes at the end of each chapter give you access to the videos in French, that are part of the MOOC. English subtitles are available for all the videos. In the early stages of learning biological crystallography, the digital version of this book can therefore be a particularly useful companion. - Marie-Hélène Le Du, Pierre Legrand, Serena Sirigu, Sylvain Ravy - EDP Sciences & Science Press Introduction to biological crystallography link: https://bit.ly/3zBC6p5
|
|
Scooped by
I2BC Paris-Saclay
September 23, 2024 10:22 AM
|
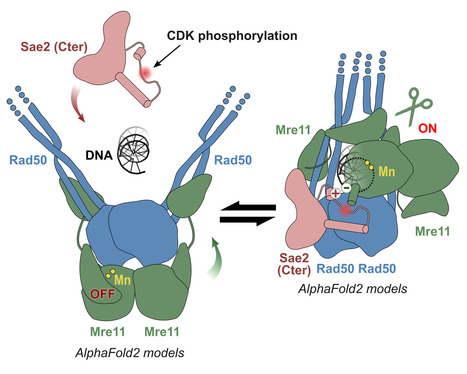
Molecular insights into the activation of Mre11-Rad50 endonuclease activity by Sae2/CtIP
Early Steps of DNA Recombination: AlphaFold2 breaks a lock. The human MRN nuclease, formed by the association of two proteins, Mre11 and Rad50, is a key player in homologous recombination and meiosis, two processes that utilize DNA break repair mechanisms. In eukaryotes, the Mre11 nuclease is equipped with a molecular lock that controls the activation of the enzyme: its activation is triggered during certain phases of the cell cycle (entry into G2, when DNA is duplicated to allow for the recombination process), when the lock is phosphorylated. This locking protein is called CtIP in humans and Sae2 in the yeast Saccharomyces cerevisiae. It is a protein with a largely disordered structure, which remained a significant challenge for structural characterization for a number of years. In a study published in Molecular Cell, teams from I2BC, the Curie Institute, and IRB shed light on how the human MRN nuclease functions. The I2BC team modeled the structure of the yeast MRN complex using the AlphaFold2 algorithm. Interestingly, the program hesitates when modeling between two states: inactive (auto-inhibited) and active. Notably, the addition of phosphorylated Sae2 favors the active conformation in AlphaFold2’s predictions: Sae2 appears to unlock the MRN complex by establishing a synergistic set of interactions centered on the phosphorylation of a serine residue in Sae2. The structural predictions obtained with AlphaFold2, supported by in vitro and in vivo experiments, highlight that the phosphorylated Sae2/CtIP protein creates a network of interactions with MRN that promotes the release of its auto-inhibition. All these findings illustrate how an apparently disordered protein can lift the auto-inhibition of a nuclease and thus control the switch between different repair pathways, which is important in humans for cell cycle progression and meiosis. More information: https://www.sciencedirect.com/science/article/pii/S1097276524004428?via%3Dihub Contact: Raphaël GUEROIS raphael.guerois@i2bc.paris-saclay.fr
|
|
Scooped by
I2BC Paris-Saclay
July 2, 2024 10:31 AM
|
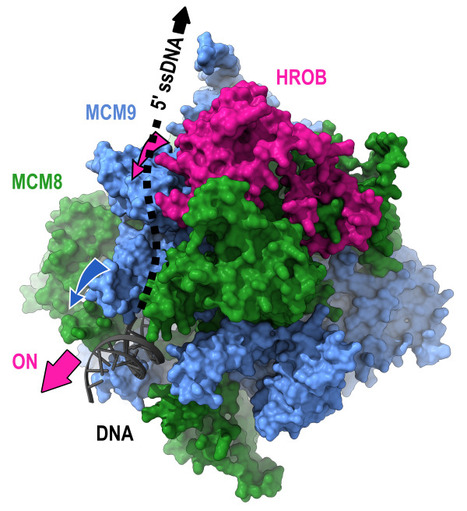
Mechanism of DNA unwinding by MCM8-9 in complex with HROB
Researchers from the AMIG team (I2BC department), in collaboration with the IRB (Switzerland), have modeled the interaction between HROB and the helicases MCM8-MCM9, some mutations of which predispose individuals to infertility or cancer. They demonstrate that HROB promotes the catalytic activity of the MCM8-MCM9 complex but does not play a role in its recruitment or stability. The proteins MCM8 and MCM9 have recently been discovered as involved in multiple processes, normal and pathological, related to DNA replication, meiosis, homologous recombination, and mismatch repair. These proteins are helicases that have the ability to move along DNA and separate the two DNA strands. Variants of these proteins may predispose carriers to disorders such as infertility and cancer. In 2019, a third partner, HROB, was identified as associated with these two helicases without its mechanisms of action being understood. Since HROB is capable of interacting with DNA, three functional hypotheses can be considered:
1) HROB participates in the recruitment of helicases on DNA,
2) HROB stabilizes the assembly of MCM8 and MCM9 on DNA,
3) HROB promotes the catalytic activity of pre-assembled helicases. In a study published in the journal Nature Communications, researchers from the AMIG team at the Institute of Integrative Biology of the Cell (I2BC) collaborated with a team from the Institute of Research in Biomedicine (IRB) in Switzerland to establish different structural models of the MCM8-MCM9-HROB complex. Guided by these models, a set of simple and compensatory mutations allowed the dissection of the functional role of HROB. The teams showed that only the third hypothesis was correct and that HROB is an essential factor for activating the MCM8-MCM9 helicase but not for its recruitment or stability. The structural model suggests that by transiently altering the conformation of a MCM9 subunit, HROB stimulates the translocation of the helicase along the DNA. Based on this model, it was even possible to design mutations that increase the efficiency of the helicase in the presence of HROB. More information: https://www.nature.com/articles/s41467-024-47936-8 Contact: Raphaël Guerois raphael.guerois@i2bc.paris-saclay.fr
|
|
Scooped by
I2BC Paris-Saclay
April 15, 2024 9:03 AM
|
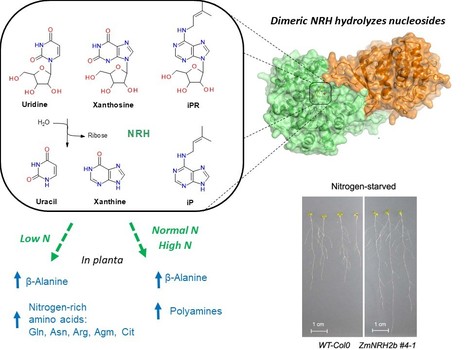
Plant nucleoside N-ribohydrolases: riboside binding and role in nitrogen storage mobilization
Plants can benefit from fast nucleoside breakdown Cells save their energy during nitrogen starvation by selective autophagy of ribosomes and degradation of RNA to ribonucleotides and nucleosides. Nucleosides are hydrolyzed by nucleoside N-ribohydrolases (nucleosidases, NRHs). Subclass I of NRHs preferentially hydrolyzes the purine ribosides while subclass II is more active towards uridine and xanthosine. Here, we performed a crystallographic and kinetic study to shed light on nucleoside preferences among plant NRHs followed by in vivo metabolomic and phenotyping analyses to reveal the consequences of enhanced nucleoside break-down. We report the crystal structure of Zea mays NRH2b (subclass II) and NRH3 (subclass I) in complexes with the substrate analog forodesine. Purine and pyrimidine catabolism are inseparable because nucleobase binding in the active site of ZmNRH is mediated via a water network and is thus unspecific. Dexamethasone-inducible ZmNRH overexpressor lines of Arabidopsis thaliana, as well as double nrh knockout lines of moss Physcomitrium patents, reveal a fine control of adenosine in contrast to other ribosides. ZmNRH overexpressor lines display an accelerated early vegetative phase including faster root and rosette growth upon nitrogen starvation or osmotic stress. Moreover, the lines enter the bolting and flowering phase much earlier. We observe changes in the pathways related to nitrogen-containing compounds such as β-alanine and several polyamines, which allow plants to reprogram their metabolism to escape stress. Taken together, crop plant breeding targetting enhanced NRH-mediated nitrogen recycling could therefore be a strategy to enhance plant growth tolerance and productivity under adverse growth conditions. more information: https://onlinelibrary.wiley.com/doi/10.1111/tpj.16572 contact: Solange Morera solange.morera@i2bc.paris-saclay.fr
|
|
Scooped by
I2BC Paris-Saclay
April 15, 2024 8:44 AM
|
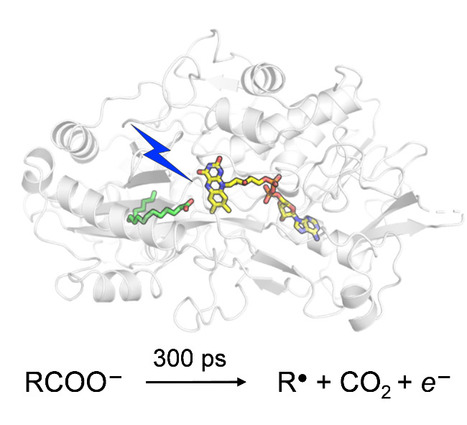
Catalytic Mechanism of Fatty Acid Photodecarboxylase: on the Detection and Stability of the Initial Carbonyloxy Radical Intermediate
Quality trumps quantity: photoenzymatic decarboxylation of fatty acids does indeed occur in 300 ps. Despite the dismissal by our "slightly overproductive" competitors, citing it as 30x slower, our original findings stand strong! In fatty acid photodecarboxylase (FAP), light-induced formation of the primary radical product RCOO● from fatty acid RCOO– occurs in 300 ps, upon which CO2 is released quasi-immediately. Based on the hypothesis that aliphatic RCOO●(spectroscopically uncharacterized because unstable) absorbs in the red similarly to aromatic carbonyloxy radicals such as 2,6-dichlorobenzoyloxy radical (DCB●), much longer-lived linear RCOO● has been suggested recently. We performed quantum chemical reaction pathway and spectral calculations. Thesecalculations are in line with the experimental DCB● decarboxylation dynamics and spectral properties and show that in contrast to DCB●, aliphatic RCOO● radicals a) decarboxylate with a very low energetic barrier and on the timescale of a few ps and b) exhibit little red absorption. A time-resolved infrared spectroscopy experiment confirms very rapid, <<300 ps RCOO● decarboxylation in FAP. We argue that this property is required for the observed high quantum yield of hydrocarbons formation by FAP. more information: https://onlinelibrary.wiley.com/doi/10.1002/anie.202401376 contact: Pavel MULLER <pavel.muller@i2bc.paris-saclay.fr>
|
|
Scooped by
I2BC Paris-Saclay
April 15, 2024 8:31 AM
|
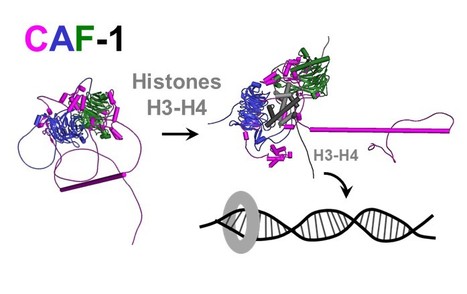
Epigenetics: combining flexible and rigid regions into a single structure to ensure genome replication and stability
The AMIG team at I2BC, in collaboration with teams from the Institut Curie and the Synchrotron Soleil, shows that the CAF-1 protein combines in its spatial organization flexible regions and rigid modules to deposit histones on DNA and effectively couple this process to DNA synthesis. Genome and epigenome integrity in eukaryotes depends on the proper coupling of histone deposition with DNA synthesis. This process relies on the evolutionary conserved histone chaperone CAF-1 for which the links between structure and functions are still a puzzle. While studies of the Saccharomyces cerevisiae CAF-1 complex enabled to propose a model for the histone deposition mechanism, we still lack a framework to demonstrate its generality and in particular, how its interaction with the polymerase accessory factor PCNA is operating. Here, we reconstituted a complete SpCAF-1 from fission yeast. We characterized its dynamic structure using NMR, SAXS and molecular modeling together with in vitro and in vivo functional studies on rationally designed interaction mutants. Importantly, we identify the unfolded nature of the acidic domain which folds up when binding to histones. We also show how the long KER helix mediates DNA binding and stimulates SpCAF-1 association with PCNA. Our study highlights how the organization of CAF-1 comprising both disordered regions and folded modules enables the dynamics of multiple interactions to promote synthesis-coupled histone deposition essential for its DNA replication, heterochromatin maintenance, and genome stability functions. More information: https://elifesciences.org/articles/91461 Contact: Françoise Ochsenbein francoise.ochsenbein@i2bc.paris-saclay.fr
On les trouve chez les animaux comme chez les plantes, les bactéries ou les virus. Elles permettent le battement régulier de votre cœur, la digestion de votre dernier repas, maintiennent votre corps à une température constante, participent à la lutte contre les infections qui vous menacent et bien plus encore... Les protéines sont partout où la vie existe et la science les étudie depuis des décennies, mais elles sont si nombreuses et si diverses que leur monde reste encore à explorer. Nous vous invitons à découvrir ces recherches passionnantes en compagnie de Marie-Hélène Le Du, chercheuse CEA à l'Institut de Biologie Intégrative de la Cellule – I2BC (DRF/Joliot/I2BC). RDV le mardi 30 janvier 2024 à 20h. Entrée libre sous réserve de places disponibles (le nombre de places est limité à 280). Vous pourrez aussi suivre cette conférence en direct sur la chaîne Youtube du CEA.
Via Life Sciences UPSaclay
|
|
Scooped by
I2BC Paris-Saclay
December 27, 2023 7:50 AM
|
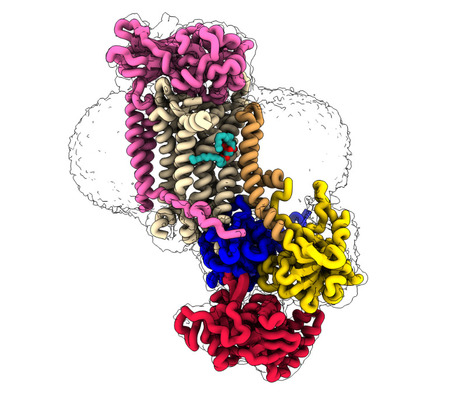
CryoEM studies of the human lipid transporter ATP8B1 involved in intrahepatic cholestasis
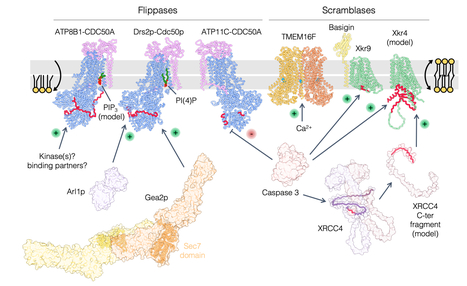
The structural characterization of the catalytic cycle of the lipid transporter ATP8B1 by cryoEM reveals fundamental aspects of its substrate specificity and of its regulation. Lipids are the main constituents of our cell membranes, which are formed as lipid bilayers. The distribution of lipids is far from uniform; it is asymmetric, with different lipid compositions in the inner and outer leaflet. This asymmetry is essential for a variety of cellular functions, from maintaining membrane homeostasis to enabling cell signaling and numerous other physiological processes including apoptosis and coagulation. P4-ATPases, also known as flippases, are key players in creating and maintaining this lipid asymmetry. These enzymes actively transport lipids from the outside (exoplasmic) leaflet to the inside (cytosolic) leaflet coupled to ATP hydrolysis and ensure the proper distributions of lipids. The ATP8B1-CDC50A flippase complex in particular, has been the subject of the current study, is involved in a rare inherited liver disease called intrahepatic cholestasis. The function of ATP8B1 lipid flippase is critical for the regulation of bile production, a vital substance in our digestive system, but the direct link within bile producing liver cells remains unknown. Additionally, recent studies have spotlighted the relevance of genetic variants in the regulatory segment of the ATP8B1 gene as a strong genetic marker for Alzheimer’s resilience. In the new study, the research team employed state-of-the-art cryo-electron microscopy techniques to capture nine different states associated with the lipid transport and determine structures at 2.4 to 3.1 Å overall resolution for these conformations. These structural insights, combined with functional and computational studies, reveal the inner workings of the human flippase ATP8B1-CDC50A complex and also resolves earlier discrepancies about the ATP8B1 transport substrates. Additionally, this work also provides key understanding about fine regulation by specific regulatory lipids known as phosphoinositides and by its autoinhibition mechanism. This work was recently awarded ”article of the month” by the Société Française de Biochimie et Biologie Moléculaire (SFBBM) in December 2023. More information: https://www.nature.com/articles/s41467-023-42828-9 Contact: Thibaud DIEUDONNE <thibaud.dieudonne@i2bc.paris-saclay.fr>
|
|
Scooped by
I2BC Paris-Saclay
December 1, 2023 8:44 AM
|
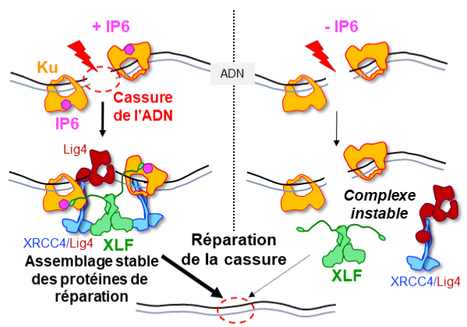
IP6 : an endogenous small molecule which promotes DNA repair.
Structural studies by X-ray cristallography and CryoEM combined with cellular analyses show the stabilizing role of IP6 in assembly of DNA double-strand break repair in human. IP6, inositol hexaphosphate or phytic acid, is a product of cellular metabolism that binds to numerous proteins whose functions it regulates. In an article published in the journal Nucleic Acids Research, scientists from I2BC (UMR 9198, CNRS/CEA/UPSaclay, Gif-sur-Yvette), from lPBS (CNRS/Université Paul Sabatier) and from Institut for Structural and Chemical Biology (University of Leicester, UK) reveal how IP6 stabilizes the assembly of a complex responsible for DNA break repair in humans. Many cancer therapies induce DNA double-strand breaks. A DNA double-strand break represents the most serious type of DNA damage to a cell, since it amounts to splitting a chromosome in two. The resulting chromosome fragments can be lost or lead to translocations if they are not quickly rejoined. Unrepaired breaks most often lead to cell death, a property exploited to eradicate tumor cells during radiotherapy or certain chemotherapies. Yet DNA double-strand break repair pathways exist, and their performance in tumors determines the efficacy of these therapies. The dominant system for repairing DNA double-strand breaks in human cells is Non-Homologous End Joining, or NHEJ, initiated by the Ku protein, which rapidly encircles the ends of the break and acts as a hub for the other proteins needed to weld these ends together. Using two techniques for studying proteins at atomic scale (crystallography and cryo-electron microscopy), the scientists discovered how a small molecule produced by the body and sometimes used as a dietary supplement, inositol-hexaphosphate (IP6) or phytic acid, binds to the Ku protein. The scientists then analyzed the effect of IP6 on break repair in human cells. They modified the Ku protein to block its binding to IP6, and thus understand how the latter acts. The Ku mutants revealed that the presence of IP6 on the Ku protein stimulates its binding to the XLF protein, an essential step in efficiently welding the DNA break (see the model below). IP6 thus plays a key role in stabilizing the large complex of NHEJ proteins required for DNA break repair. This fundamental work resolves the question raised two decades ago regarding the role of IP6 in DNA double-strand break repair. It also opens up therapeutic prospects by identifying a new region on the Ku protein that could be targeted by small molecules to block DNA break repair in tumor cells. More information: https://academic.oup.com/nar/advance-article/doi/10.1093/nar/gkad863/7327077?login=false Contact: Jean-Baptiste CHARBONNIER <jb.charbonnier@i2bc.paris-saclay.fr>
|
|
Scooped by
I2BC Paris-Saclay
October 23, 2023 4:58 AM
|
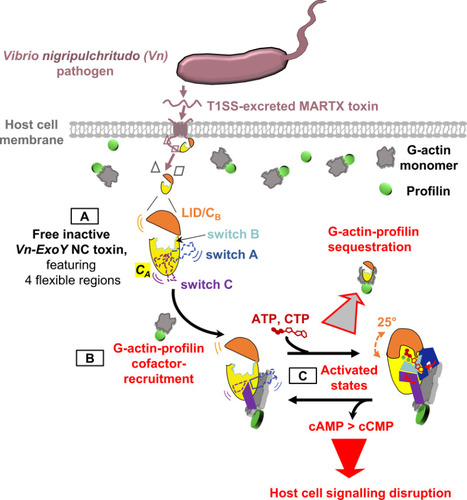
Functional and structural insights into the multi-step activation and catalytic mechanism of bacterial ExoY nucleotidyl cyclase toxins bound to actin-profilin
We reveal unprecedented mechanistic structural details of how the active site of bacterial Nucleotidyl Cyclase toxins is sequentially remodeled by cofactor and substrate binding and how they can accommodate both purine or pyrimidine nucleotides as substrate. Nucleotidyl cyclase (NC) enzymes are important ubiquitous enzymes that catalyze the production of cyclic nucleotides, crucial second messengers involved in numerous signaling pathways. ExoY-like toxins are bacterial virulence factors produced by Gram-negative γ- and β-proteobacteria. They belong to a broader family of bacterial NC toxins. When delivered into eukaryotic cells, NC toxins alter host cell signalling by overproducing both cyclic purine (cAMP, cGMP) and pyrimidine (cCMP, cUMP) nucleotides. To become potent NC enzymes inside host cells, they bind to specific host cofactors. The molecular and mechanistic details underlying the activation and catalytic specificities of NC toxins are only partially understood. ExoY NC toxins utilize either polymerized or monomeric (G-actin) actin as cofactor.
Here, we (a team at I2BC collaborating with scientists at ICSN, CNRS, UPS, Gif-Sur-Yvette, and at Institute Pasteur, Paris) investigated ExoY-like NCs that are selective for G-actin. Our in vitro investigations first revealed the physiological cofactor at work within host eukaryotic cells. These ExoYs activate by capturing and sequestering the cytoskeletal G-actin-profilin complex, disrupting its regulatory functions in the dynamic assembly of actin (Fig. 1, steps B-C).
Using X-ray crystallography at SOLEIL synchrotron, we have intercepted several structural snapshots along the ExoY activation pathway by G-actin-profilin. These structural data reveal unprecedented mechanistic details of how the active site of all NC toxins undergoes progressive remodeling upon cofactor and substrate binding (Fig. 1, step C). They reveal how NC toxins can accommodate purine or pyrimidine nucleotides as substrates and clarify crucial aspects of their catalytic purinyl and pyrimidinyl cyclase reaction. These structural insights into the multi-step activation of NC toxins may open new avenues to inhibit specifically this class of toxic bacterial NCs. More information: https://journals.plos.org/plospathogens/article?id=10.1371/journal.ppat.1011654 Contact: Louis RENAULT <louis.renault@i2bc.paris-saclay.fr>
|
|
Scooped by
I2BC Paris-Saclay
October 23, 2023 4:20 AM
|
Cyclope conference "Proteins are everywhere"
This conference is postponed to a later date. The next Cyclope conference organized by the CEA will be presented by Marie-Hélène Le DU at the INSTN at 8 p.m on the 24th of October 2023. The headline: “Proteins are everywhere". They are found in animals as well as plants, bacteria or viruses. They allow the regular beating of your heart, the digestion of your last meal, maintain your body at a constant temperature, participate in the fight against infections that threaten you and much more...
Proteins are everywhere life exists and science has studied them for decades, but they are so numerous and so diverse that their world has yet to be explored. We invite you to discover this exciting research in the company of Marie-Hélène Le Du, CEA researcher at the Institute of Integrative Cell Biology (DRF/Joliot/I2BC), with the participation of Rémi Ruedas, a doctoral student at I2BC.
|
|
Scooped by
I2BC Paris-Saclay
September 20, 2023 5:17 AM
|
Multiscale Transient Absorption Study of the Fluorescent Protein Dreiklang and Two Point Variants Provides Insight into Photoswitching and Nonproductive Reaction Pathways
Photoswitching and non-productive reaction pathways of the fluorescent protein 'Dreiklang' deciphered in a comprehensive time-resolved spectroscopic study. Dreiklang is a reversibly photoswitchable fluorescent protein used as a probe in advanced fluorescence imaging. It undergoes a unique and still poorly understood photoswitching mechanism based on the reversible addition of a water molecule to the chromophore. We report the first comprehensive study of the dynamics of this reaction by transient absorption spectroscopy from 100 fs to seconds in the original Dreiklang protein and its two point variants. The picture that emerges from our work is that of a competition between photoswitching and nonproductive reaction pathways. We found that photoswitching had a low quantum yield of 0.4%. It involves electron transfer from a tyrosine residue (Tyr203) to the chromophore and is completed in 33 ns. Nonproductive deactivation pathways comprise recombination of a charge transfer intermediate, excited-state proton transfer from the chromophore to a histidine residue (His145), and decay to the ground state via micro-/millisecond-lived intermediates. More information: https://pubs.acs.org/doi/10.1021/acs.jpclett.3c00431 Contact: Pavel MULLER <pavel.muller@i2bc.paris-saclay.fr>
|
|
Scooped by
I2BC Paris-Saclay
July 19, 2023 10:22 AM
|
Phosphatidylserine transport in cell life and death
Lipids beyond membrane building blocks: recent advances in our understanding of phosphatidylserine distribution in eukaryotic cells and its role in infectious diseases. A prominent feature of cell membranes is the non-uniform distribution of the lipids they are made of. This is particularly true for the negatively charged glycerophospholipid phosphatidylserine (PS) which is enriched in the plasma membrane and in late secretory/endocytic compartments. In addition to the uneven distribution of PS between membranes, the transbilayer distribution (the enrichment of a given lipid in one membrane leaflet over the other) of PS is tightly regulated, as it is largely confined to the cytosolic leaflet of membranes, where it can mediate the recruitment of a diverse set of proteins. As such, PS controls a myriad of cell signaling pathways as well as membrane trafficking events. In some specific conditions, PS may however be ‘flipped’ toward the external leaflet of the plasma membrane, where it can act as an ‘eat-me’ signal for engulfment of apoptotic cells by macrophages or to promote synaptic pruning, which consists in the removal of synapses to establish proper connections during brain development. The exquisitely tailored transbilayer PS distribution is also crucial in host-pathogen interactions, as it is exploited by various intracellular pathogens to infect cells. In this review, we highlight recent findings on nonvesicular PS transport between membranes by lipid-transfer proteins at membrane contact sites, on PS flip-flop between membrane leaflets by lipid flippases and scramblases, and we discuss how perturbation of PS distribution can lead to disease and the role of PS in viral infection. More information: https://www.sciencedirect.com/science/article/pii/S0955067423000418?via%3Dihub Contact: Guillaume Lenoir <guillaume.lenoir@i2bc.paris-saclay.fr>
|
|
|
Scooped by
I2BC Paris-Saclay
October 17, 2024 5:43 AM
|
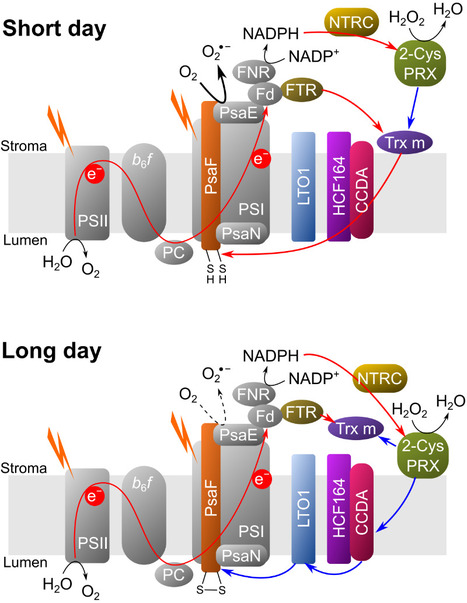
A complex and dynamic redox network regulates oxygen reduction at photosystem I in Arabidopsis
A system of redox enzymes facilitates a series of reactions that regulate oxygen reduction at photosystem I in plants. In a study published in Plant Physiology, scientists from I2BC and IPS2 studied the redox regulation of superoxide production at the photosynthetic electron transfer level (pseudocyclic flow). Plants must optimise their molecular strategies to adapt to the light conditions of their environment. Reactive oxygen species (ROS) can play a role as signalling molecules but can also be potentially harmful. To manage light-induced ROS and maintain proper photosynthetic function, thiol-dependent redox enzymes play a crucial role in the redox regulation of the chloroplast. It has been shown that the main redox enzymes involved in controlling the ROS generated during photosynthesis are m-type thioredoxins, NADPH-dependent reductase C (NTRC) and peroxiredoxin 2-Cys. The interaction of these enzymes functions as a redox regulatory network, where 2-Cys PRX can be reduced by NTRC and Trx but can also re-oxidise the reduced thioredoxins, thus providing a system for rapid acclimatisation to changes in the light regime. The researchers showed that the membrane localisation of Trx m and NTRC varies as a function of photoperiod. In addition, their results show that it is the PSI itself that is redox regulated. The new results of this study make it possible to generate a new model of PSI redox regulation. This model may guide further research into the redox regulation of alternative electron transport pathways under conditions of fluctuating light or abiotic stress. More information: DOI: 10.1093/plphys/kiae501 Contact: Anja KRIEGER-LISZKAY anja.liszkay@i2bc.paris-saclay.fr
|
|
Scooped by
I2BC Paris-Saclay
July 26, 2024 8:24 AM
|
BRCA2 stabilizes DMC1 nucleoprotein filaments in meiosis
To repair programmed double-strand breaks in meiosis, the DNA repair protein BRCA2 binds to the recombinase DMC1 either monomeric or assembled on single-stranded DNA through two different interfaces, and stabilizes DMC1 nucleoprotein filaments. The BReast CAncer type 2 susceptibility protein (BRCA2), a tumor suppressor mutated in breast, ovarian and prostate cancers, plays a major role in the repair of DNA double-strand breaks by homologous recombination, both in somatic cells and during meiosis. BRCA2 interacts with the ubiquitous recombinase RAD51 , as well as the meiotic recombinase DMC1, and facilitates their loading at double-strand break sites. BRCA2 interacts with these recombinases via FxxA and FxPP motifs (called A and P motifs, respectively). In a study published recently in the journal Nucleic Acids Research, scientists from the INTGEN team at the I2BC (Université Paris Saclay, CEA, CNRS, Gif-sur-Yvette) and the PROXIMA-1 beamline at the SOLEIL synchrotron (Université Paris Saclay) solved the crystal structure of the complex between a BRCA2 fragment containing a P motif (PhePP) and the DMC1 protein. Together with the team of A. Zelensky and R. Kanaar (Erasmus Medical Center, Rotterdam), they showed that A and P motifs bind to distinct sites on the ATPase domain of recombinases. The P motif interacts with a site that is accessible in the octamers of DMC1 and the nucleoprotein filaments formed by DMC1 loaded onto single-stranded DNA. Furthermore, in collaboration with scientists from the Institut Gustave Roussy (Université Paris Saclay), they revealed that this interaction involves two adjacent DMC1 protomers, thereby increasing the stability of the nucleoprotein filaments. These results help to explain why the region encoded by exons 12 to 14 of the BRCA2 gene (the PhePP motif being encoded by exon 14) is essential during meiotic homologous recombination in mice (work by the teams of A. Zelensky and W. Baarends, Erasmus Medical Center, Rotterdam, Netherlands). More information: https://doi.org/10.1093/nar/gkae452 Contact: Simona MIRON <simona.miron@i2bc.paris-saclay.fr> & Sophie ZINN <sophie.zinn@i2bc.paris-saclay.fr>
|
|
Scooped by
I2BC Paris-Saclay
April 15, 2024 9:06 AM
|
Physics and Biological Systems 2024
Mark your calendars for Jun. 26-28, 2024! Join us at Ecole Polytechnique in Palaiseau for the 7th International Conference on Physics and Biological Systems, where top-notch scientists converge to explore the dynamic interplay between physics and life sciences. The 7th International Conference on Physics and Biological Systems will be held on Jun. 26-28 2024 at Ecole Polytechnique in Palaiseau, in the south of Paris. It aims to bring together a broad range of physical and life scientists working at the interface between the two disciplines around in-depth talks by first-rate international speakers. Attendance will be limited to 200 participants. We look forward to welcoming you in Palaiseau! more information: https://www.lptms.universite-paris-saclay.fr/physbio2024/ contact: Pavel MULLER <pavel.muller@i2bc.paris-saclay.fr>
|
|
Scooped by
I2BC Paris-Saclay
April 15, 2024 8:50 AM
|
A high-end Glacios2 cryo-electron microscope at I2BC
A Glacios2 200 kV has been delivered to I2BC, heralding, together with a Titan Krios 300 kV at synchrotron SOLEIL, a new era for cryo-electron microscopy in the Paris-Saclay area Cryo-electron microscopy (cryo-EM) has become an indispensable tool in structural biology, enabling researchers to unravel the complexities of biological macromolecules and understand their roles in various cellular processes and diseases. These last ten years the field has undergone a revolution, with technical advances that have made atomic resolution reconstructions of large macromolecular complexes possible (the so-called 'resolution revolution'), I2BC received the latest generation 200kV microscope, Glacios2, on March 27, 2024. Joint operation is planned with the 300kV Titan Krios also just delivered at synchrotron SOLEIL. This will allow researchers to study structures of living machines at work in fine detail, both for purified and/or reconstituted complexes and in cells. These two cryo-electron microscopes are gathering scientists from all over the Paris-Saclay area, sparking further interest in future developments at the interface beween structural and cellular biology. contact: Stéphane Bressanelli (plt-cryoem@i2bc.paris-saclay.fr)
|
|
Scooped by
I2BC Paris-Saclay
April 15, 2024 8:33 AM
|
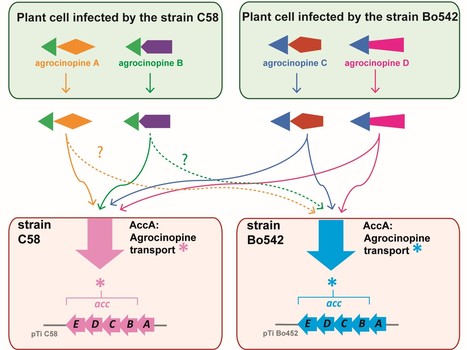
A highly conserved ligand-binding site for AccA transporters of antibiotic and quorum-sensing regulator in Agrobacterium leads to a different specificity
The highly conserved ligand binding site of the AccA transporters of antibiotic and quorum-sensing regulator in Agrobacterium is not linked to a conserved specificity. Plants genetically modified by the pathogenic Agrobacterium strain C58 synthesize agrocinopines A and B, whereas those modified by the pathogenic strain Bo542 produce agrocinopines C and D. The four agrocinopines (A, B, C and D) serve as nutrients by agrobacteria and signaling molecule for the dissemination of virulence genes. They share the uncommon pyranose-2-phosphate motif, represented by the L-arabinopyranose moiety in agrocinopines A/B and the D-glucopyranose moiety in agrocinopines C/D, also found in the antibiotic agrocin 84. They are imported into agrobacterial cytoplasm via the Acc transport system, including the solute-binding protein AccA coupled to an ABC transporter. We have previously shown that unexpectedly, AccA from strain C58 (AccAC58) recognizes the pyranose-2-phosphate motif present in all four agrocinopines and agrocin 84, meaning that strain C58 is able to import agrocinopines C/D, originating from the competitor strain Bo542. Here, using agrocinopine derivatives and combining crystallography, affinity and stability measurements, modeling, molecular dynamics, in vitro and vivo assays, we show that AccABo542 and AccAC58 behave differently despite 75% sequence identity and a nearly identical ligand binding site. Indeed, strain Bo542 imports only compounds containing the D-glucopyranose-2-phosphate moiety, and with a lower affinity compared to strain C58. This difference in import efficiency makes C58 more competitive than Bo542 in culture media. We can now explain why Agrobacterium/Allorhizobium vitis strain S4 is insensitive to agrocin 84, although its genome contains a conserved Acc transport system. Overall, our work highlights AccA proteins as a case study, for which stability and dynamics drive specificity. More information: https://portlandpress.com/biochemj/article-abstract/481/2/93/233802/A-highly-conserved-ligand-binding-site-for-AccA?redirectedFrom=fulltext Contact: Solange Morera solange.morera@i2bc.paris-saclay.fr
|
|
Scooped by
I2BC Paris-Saclay
February 19, 2024 10:12 AM
|
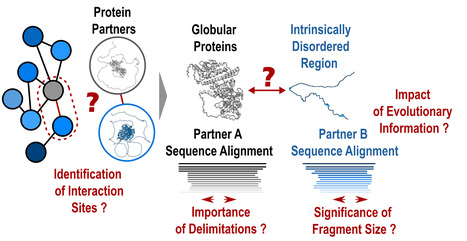
Protein-protein interactions: how to push forward the limits of the revolutionary AlphaFold2 programme?
AlphaFold2 is revolutionising protein structure prediction and structural biology practices. However, it may prove less effective for certain protein assemblies, particularly when they depend on intrinsically disordered regions. In an article published in Nature Communications, researchers from the I2BC show that applying a fragmentation strategy to the protein partners of such assemblies very significantly improves AlphaFold2’s prediction capacity. Mapping protein-protein interaction networks is essential for understanding the dynamics of cellular functions and their cross-regulation. Precise knowledge of interaction sites makes it possible to specifically perturb the proteins in these networks and understand the synergies and competitions that ensure cell function. Unfortunately, a great amount of structural information is still lacking to provide a detailed understanding of the organisation of interaction networks. The AlphaFold2 artificial intelligence programme has demonstrated a remarkable ability to predict the structures of protein assemblies that have co-evolved over long time scales. Its performance remained poorly characterised for assemblies involving intrinsically disordered regions, which often mediate transient and dynamic interactions during evolution. In a study published in the journal Nature Communications, researchers from the AMIG team at the Institut de Biologie Intégrative de la Cellule – I2BC (CNRS/CEA/UPSaclay, Gif-sur-Yvette) have shown that AlphaFold2 performs poorly if large disordered regions are used directly for prediction (40% success rate). A protein fragmentation strategy was found to be particularly well adapted to predicting the interfaces between folded domains and small protein motifs that fold on contact with the partner. Applied on a large scale using the Jean Zay HPC infrastructure on more than 900 complexes, this strategy achieved a success rate of almost 90%, a very encouraging result for the systematic screening of protein interaction networks. Nevertheless, the study calls for vigilance with regard to the risks of detecting false positives, which will be at the heart of future developments in artificial intelligence strategies such as AlphaFold2. More information: https://www.nature.com/articles/s41467-023-44288-7 Contact: Jessica ANDREANI and Raphaël GUEROIS <jessica.andreani@i2bc.paris-saclay.fr> <raphael.guerois@i2bc.paris-saclay.fr>
|
|
Scooped by
I2BC Paris-Saclay
December 27, 2023 8:28 AM
|
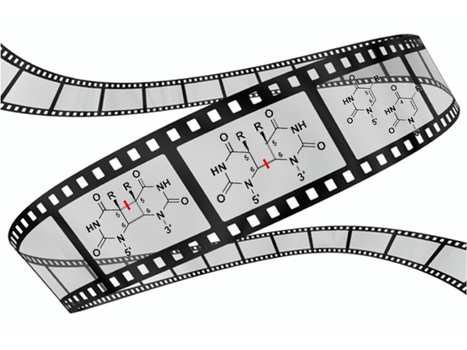
Visualizing the DNA repair process by a photolyase at atomic resolution
Molecular movies? No problem for time-resolved serial femtosecond X-ray crystallography! We have obtained 18 snapshots visualizing the whole DNA repair by a photolyase: from the initial electron transfer to the active site recovery and DNA reannealing. Photolyases, a ubiquitous class of flavoproteins, use blue light to repair DNA photolesions. In this work, we determined the structural mechanism of the photolyase-catalyzed repair of a cyclobutane pyrimidine dimer (CPD) lesion using time-resolved serial femtosecond crystallography (TR-SFX). We obtained 18 snapshots that show time-dependent changes in four reaction loci. We used these results to create a movie that depicts the repair of CPD lesions in the picosecond-to-nanosecond range, followed by the recovery of the enzymatic moieties involved in catalysis, completing the formation of the fully reduced enzyme-product complex at 500 nanoseconds. Finally, back-flip intermediates of the thymine bases to reanneal the DNA were captured at 25 to 200 microseconds. Our data cover the complete molecular mechanism of a photolyase and, importantly, its chemistry and enzymatic catalysis at work across a wide timescale and at atomic resolution. More information: https://www.science.org/doi/10.1126/science.add7795 Contact: Pavel MULLER <pavel.muller@i2bc.paris-saclay.fr>
|
|
Scooped by
I2BC Paris-Saclay
December 1, 2023 8:46 AM
|
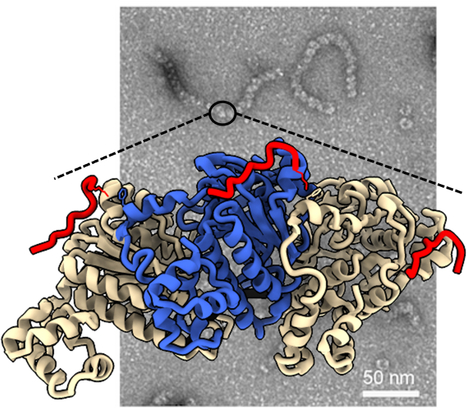
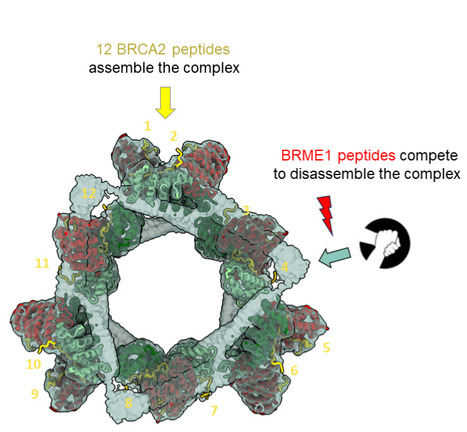
The large ring-shaped BRCA2-HSF2BP complex is disassembled by BRME1, thus promoting Homologous Recombination: a cryo-EM study.
A short disordered and conserved motif of the tumor suppressor BRCA2 triggers the assembly of a large ring-shaped 24-mer of the meiotic HSF2BP protein, whereas the disordered C-terminal motif of the meiotic BRME1 dissociates this ring complex. In meiotic homologous recombination (HR), BRCA2 facilitates loading of the recombinases RAD51 and DMC1 at the sites of double-strand breaks (DSB). The HSF2BP-BRME1 complex interacts with BRCA2. Its absence causes a severe reduction in recombinase loading at meiotic DSB. We previously showed that, in somatic cancer cells ectopically producing HSF2BP, DNA damage can trigger HSF2BP-dependent degradation of BRCA2, which prevents HR. Here we report that, upon binding to BRCA2, HSF2BP forms octameric rings that are able to assemble into a large ring-shaped 24-mer. Addition of BRME1 leads to dissociation of both of these ring structures, and cancels the disruptive effect of HSF2BP on cancer cell resistance to DNA damage. It also prevents BRCA2 degradation during inter-strand DNA crosslink repair in Xenopus egg extracts. We propose that the control of HSF2BP-BRCA2 oligomerization by BRME1 ensures timely assembly of the ring complex that concentrates BRCA2 and controls its turnover, thus promoting HR. More information: https://www.science.org/doi/10.1126/sciadv.adi7352 Contact: Sophie ZINN JUSTIN <sophie.zinn@i2bc.paris-saclay.fr>
|
|
Scooped by
I2BC Paris-Saclay
October 23, 2023 5:08 AM
|
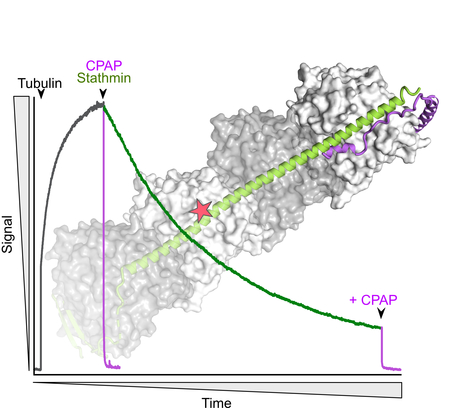
The C-terminus of stathmin-like proteins governs the stability of their complexes with tubulin
Identification of a novel mechanism for the regulation of the stathmin:tubulin interaction. Microtubule dynamics is modulated by many cellular factors including stathmin family proteins. Vertebrate stathmins sequester two ab-tubulin heterodimers into a tight complex that cannot be incorporated in microtubules. Stathmins are regulated at the expression level during development and among tissues; they are also regulated by phosphorylation. We have studied the dissociation kinetics of tubulin:stathmin assemblies in presence of different tubulin-binding proteins and identified a critical role of the C-terminus of the stathmin partner. Destabilizing this C-terminal region may represent an additional regulatory mechanism of the interaction with tubulin of stathmin proteins. More information: https://doi.org/10.1016/j.bbrc.2023.10.023 Contact: Benoît GIGANT <benoit.gigant@i2bc.paris-saclay.fr>
|
|
Scooped by
I2BC Paris-Saclay
October 23, 2023 4:50 AM
|
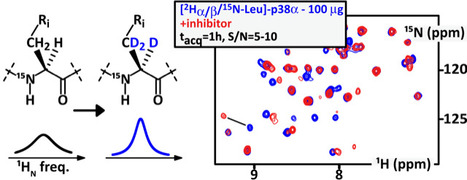
100 ug of kinase are now enough for its residue-scale NMR characterization
Kinases are exquisite drug targets, but usually too large for any NMR characterization at decent concentrations; we combined a number of innovative strategies enabling S/N~10 in 1h using less than 100 ug of protein. We aimed at designing and testing affordable strategies for improving NMR sensitivity on common-size drug targets, e.g. kinases of 40 kDa. This goes notably via novel isotope labeling schemes, which we sought to be as versatile as possible in terms of recombinant expression organism.
We evaluated the benefits of the deuteration on alpha- and beta-positions of amino acids for a typical middle size protein domain, namely the model 40 kDa-large kinase p38α.
We used cystathionine gamma-synthase (and new high-performance mutants thereof) to execute position-specific deuteration of free amino acids by H/D exchange. Then, we employed cell-free expression in bacterial extracts to avoid any scrambling and back-protonation of the tested isotopically labelled amino acids (Ala, Leu, Lys, Ser, Asp, Glu, Gly).
Altogether, these combined strategies yielded a ten-fold sensitivity increase, as compared to the former classical NMR approaches. It can be applied using recombinant expression in cell-free systems and in eukaryotic cells. This allows recording residue-resolved solution 1H-15N NMR spectra of 100 μg of purified p38α in one hour with S/N∼10, and a residue-specific characterization of drug binding.
We want to apply this approach for in-cell NMR studies in the future. More information: https://www.sciencedirect.com/science/article/pii/S2666441023000341?via%3Dihub Contact: Theillet Francois-Xavier <francois-xavier.theillet@i2bc.paris-saclay.fr>
|
|
Scooped by
I2BC Paris-Saclay
September 20, 2023 8:48 AM
|
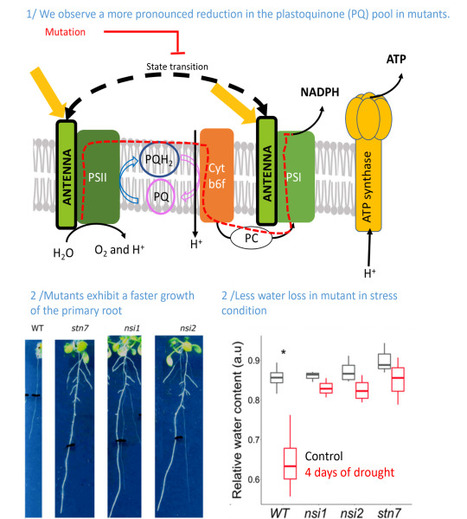
Identification of a new pathway potentially involved in plant resistance to drought
Light capture by the chlorophyll containing antenna and the reduction state of the photosynthetic electron transport chain affect root development. Identifying traits that exhibit improved drought resistance is highly important to cope with the challenges of predicted climate change. We investigated the response of state transition mutants to drought. Compared with the wild type, state transition mutants were less affected by drought. Photosynthetic parameters in leaves probed by chlorophyll fluorescence confirmed that mutants possess a more reduced plastoquinone (PQ) pool, as expected due to the absence of state transitions. Seedlings of the mutants showed an enhanced growth of the primary root and more lateral root formation. The photosystem II inhibitor 3-(3,4-dichlorophenyl)-1,1-dimethylurea, leading to an oxidised PQ pool, inhibited primary root growth in wild type and mutants, while the cytochrome b6 f complex inhibitor 2,5-dibromo-3-methyl-6-isopropylbenzoquinone, leading to a reduced PQ pool, stimulated root growth. A more reduced state of the PQ pool was associated with a slight but significant increase in singlet oxygen production. Singlet oxygen may trigger a, yet unknown, signalling cascade promoting root growth. We propose that photosynthetic mutants with a deregulated ratio of photosystem II to photosystem I activity can provide a novel path for improving crop drought resistance. More information: https://pubmed.ncbi.nlm.nih.gov/37614199/ Contact: Anja KRIEGER-LISZKAY <anja.krieger-liszkay@i2bc.paris-saclay.fr>
|
|
Scooped by
I2BC Paris-Saclay
September 19, 2023 8:58 AM
|
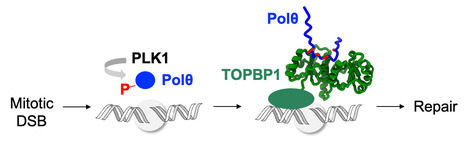
Phosphorylation of polymerase theta by PLK1 is essential for the repair of double-strand breaks in mitosis through a novel DNA repair pathway.
The DNA polymerase theta is phosphorylated by PLK1 in mitosis, binds to TOPBP1 and is recruited to double-strand breaks, in order to trigger repair through a novel DNA repair pathway, thus becoming a new target for the treatment of breast and ovarian cancers. DNA double-strand breaks (DSBs) are deleterious lesions that challenge genome integrity. In interphase, DSBs are mainly repaired by non-homologous end joining and homologous recombination. In mitosis, specific kinases inhibit these pathways through phosphorylation of essential DNA repair proteins. Here we show that one of these mitotic kinases, Polo-like kinase 1 (PLK1), activates the DNA polymerase theta (Polθ), which is then recruited through an interaction with TOPBP1 to mitotic DSBs. NMR analyses demonstrate that PLK1 phosphorylates a cluster of four serines in the central disordered region of Polθ, and that the phosphorylated motif directly interacts with the C-terminal domains of TOPBP1. The Artificial Intelligence based program AlphaFold consistently predicts that the Polθ region containing the cluster of four serines binds in a groove at the surface of the C-terminal domains of TOPBP1. Mutating these serines impairs recruitment of Polθ to mitotic DSBs and joining of the broken DNA ends. Polθ is essential for the repair of mitotic DSBs. Its role is even more crucial in cells that are deficient in homologous recombination, because these cells accumulate DSBs at the entry of mitosis, and loss of mitotic DSB repair by Polθ results in cell death. Our data explains why Polθ is synthetic lethal with homologous recombination deficiency, and reveals the critical importance of mitotic DSB repair in the maintenance of genome integrity. More information: https://www.nature.com/articles/s41586-023-06506-6 Contact: Sophie ZINN JUSTIN <sophie.zinn@i2bc.paris-saclay.fr>
|