Your new post is loading...
Your new post is loading...

|
Scooped by
I2BC Paris-Saclay
January 28, 8:19 AM
|
Crosstalk between cohesins and axis proteins determines meiotic chromosome architecture in Sordaria macrospora
New discovery: a dynamic interplay between axis proteins and cohesins ensures chromosome stability during meiosis in Sordaria. Faithful chromosome segregation during meiosis requires the coordinated action of cohesin complexes and chromosome axis proteins. How these factors interact and communicate along chromosome axes, especially during meiotic prophase I, remains however, only partially understood. Researchers of the Genome Biology Department of the I2BC investigated the functional interplay between the cohesin components and regulators (Rad21, Rec8, Wapl, Sororin, Spo76/Pds5) and two meiosis-specific axis proteins Red1 and Hop1. Analysis of multiple combinations of their corresponding null mutants and of their genetic-epistasis interactions in the fungus Sordaria macrospora revealed a hierarchical regulatory network for their recruitment and releasing. Their work uncovers an unexpected role of axis proteins Red1 and Hop1, that together with Sororin, provide stage-specific protection of Spo76/Pds5 against Wapl-mediated release. Furthermore, we identify that Spo76/Pds5 is the main target of Wapl and acts as a central guardian of kleisin stability against Slx8/STUbL-dependent proteasomal degradation. Together, these findings show that a dynamic crosstalk between axis proteins and cohesins is crucial to preserve axis integrity and to ensure accurate meiotic progression. More information: https://doi.org/10.1371/journal.pgen.1012001 Contact: Stéphanie BOISNARD stephanie.boisnard@i2bc.paris-saclay.fr
|
|
Scooped by
I2BC Paris-Saclay
January 13, 3:30 PM
|
How virulence genes reorganize the Salmonella genome
Using functional genomics in sorted Salmonella populations and high-resolution microscopy, the researchers of the I2BC show that the expression of Pathogenicity Island 1 is associated with chromatin remodeling and with the repositioning of this region toward the nucleoid periphery. Chromatin provides a universal framework for organizing and regulating genomes across the three domains of life. In bacteria, it is composed of intrinsically supercoiled DNA associated with small DNA-binding proteins known as nucleoid-associated proteins (NAPs). Their binding can induce DNA bending, bridging, coating, and/or wrapping, giving rise to distinct modes of chromatin organization.
Bacterial chromatin can exist in a repressed state (silent chromatin) or in an actively transcribed state (active chromatin). Silent chromatin is largely associated with H-NS, a xenogeneic silencer that restricts the costly expression of genes acquired by horizontal transfer. In contrast, active chromatin is densely occupied by RNA polymerase and is characterized by different levels of DNA supercoiling. However, the changes in protein occupancy and chromatin organization that accompany transitions between these two states remain poorly understood.
Researchers of the Genome Biology Department of the I2BC in collaboration with the NGS and the Imaging facility of the I2BC and the Trinity College Dublin (Ireland), have unveiled the chromatin organization of horizontally acquired regions in Salmonella enterica serovar Typhimurium, which are essential for its pathogenicity. They show that expression of Pathogenicity Island 1 (SPI-1) is accompanied by local chromatin remodeling, marked by profound changes in three-dimensional organization and protein occupancy. This remodeling is also associated with the repositioning of SPI-1 toward the nucleoid periphery.
These findings provide new insights into the interplay between xenogeneic silencing, counter-silencing mechanisms, chromatin architecture, and the evolutionary integration of acquired DNA. They also reveal a finely tuned chromatin remodeling process that minimizes the cellular cost of activating pathogenicity islands, and they establish a direct link between the linear (1D) organization of the genome and its three-dimensional (3D) folding. More information: https://www.nature.com/articles/s41467-025-67746-w https://www.insb.cnrs.fr/fr/cnrsinfo/comment-les-genes-de-virulence-reorganisent-le-genome-de-salmonella-0 Contact: Vicky Lioy virginia.lioy@i2bc.paris-saclay.fr
|
|
Scooped by
I2BC Paris-Saclay
December 8, 2025 4:54 AM
|
Selective elimination of donor bacteria enables global profiling of plasmid gene expression during conjugation
A new ED-TA method which enabled genomics investigation of plasmid establishment during conjugative transfer was developed by I2BC researchers. They showed robust induction of a subset of plasmid genes at the early stages of conjugation through single-stranded DNA promoters. Bacterial conjugation is a principal mode of horizontal gene transfer which has important life science implications including bacterial genome evolution, dissemination of genetic traits and bioengineering applications. Notably, the spread of multidrug resistance via conjugative plasmids is one of the biggest concerns in global public health. Although conjugation has been studied since 1940s and the overall procedure is widely known and well documented, molecular details of reactions that establishes a transferred plasmid in the new host cell remain elusive. In addition, there are specific regulatory mechanisms for temporal expression of plasmid genes, that are also crucial for successful conjugation as they allow timely expression and function of plasmid-encoded arsenal against host defense mechanisms. Genomics-based studies of plasmid establishment were previously hampered by the nature of conjugation which takes place within a mixture of cell populations. Essentially, they require the separation of subpopulations before the DNA/RNA extraction to avoid contamination of indistinguishable plasmid DNA/RNA from the donor. Researchers of the Genome Biology Department of the I2BC, described the development of a new method, called ED-TA, which exploits a donor mutant hypersensitive to hypoosmotic shock. ED-TA allows unprecedently quick and efficient « Elimination of Donor population for Transconjugant Analysis». Using a clinically relevant model multidrug resistant conjugative plasmid, pESBL, they elucidated its transcription profile in successful and abortive conjugation. Researchers also showed single-stranded DNA promoters allow robust induction of a subset of genes at the early stages of conjugation. As the ED-TA method is straightforward and broadly applicable, further research taking advantage of the method will shed light on important molecular mechanisms of plasmid establishment after conjugation. More information: doi.org/10.1093/nar/gkaf1299 https://www.insb.cnrs.fr/fr/cnrsinfo/une-nouvelle-methode-pour-suivre-de-pres-la-transmission-des-genes-chez-les-bacteries Contact: Yoshiharu Yamaichi yoshiharu.yamaichi@i2bc.paris-saclay.fr
|
|
Scooped by
I2BC Paris-Saclay
October 6, 2025 3:37 PM
|
New team in the Department of Genome Biology
Cécile Courret, CNRS Researcher and recipient of an ATIP-Avenir grant, has joined the Department of Genome Biology to establish her team ‘Intragenomic Conflict and Evolution’. Cécile's research focus on genetic conflicts and their impact on genome evolution. The basic principles of Mendelian inheritance state that in heterozygous individuals, two alleles have equal chances of being transmitted to the next generation. However, some genetic elements do not follow these rules. These so-called selfish genetic elements, such as transposons or meiotic drivers, bias inheritance in their own favor, often at the expense of the organism. Because they disrupt essential processes like meiosis, heterochromatin regulation, and cell division, selfish elements create a persistent conflict with the host genome. This conflict triggers an evolutionary “arms race,” in which genomes evolve defense mechanisms to counterbalance the harmful effects of these elements. Far from being rare exceptions, such conflicts are now recognized as a major force shaping genome structure and function. My research relies on the Drosophila model and combines genomic, molecular, and cytological approaches to investigate both how selfish elements perturb fundamental biological processes and how organisms respond to mitigate these disruptions. By studying systems where these conflicts are still active in natural populations, as well as the genomic signatures left by past events, Cécile's team aims to better understand how conflicts drive evolutionary innovation and shed light on the molecular basis of essential biological mechanisms. Contact: Cécile Courret, cecile.courret@i2bc.paris-saclay.fr
|
|
Scooped by
I2BC Paris-Saclay
April 29, 2025 2:50 AM
|
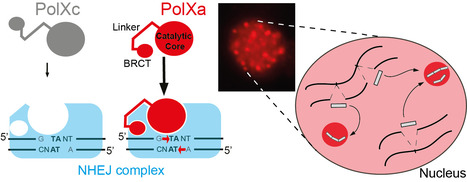
Paramecium PolX DNA polymerases keep the genome safe during programmed rearrangements
The expansion of PolX DNA polymerases in the ciliate Paramecium tetraurelia has favored the functional diversification of a subset of these enzymes, with enhanced nuclear localization in DNA repair foci during programmed DNA elimination. During its sexual cycle, Paramecium massively rearranges its genome by removing tens of thousands of internal eliminated sequences (IESs) from its developing somatic nucleus. This process involves the introduction of programmed DNA double-strand breaks (DSBs), followed by efficient DSB repair using the non-homologous end joining (NHEJ) pathway. In this system, DSB repair requires not only the core NHEJ factors Ku70/80 and Xrcc4/Lig4, but also additional enzymes that accurately process the 4-base 5′-overhangs of the broken DNA ends created at IES excision sites. In this study, we identify four orthologs of human DNA polymerase lambda in P. tetraurelia —PolXa, PolXb, PolXc, and PolXd— as key players in repairing IES excision junctions. Cells lacking these PolX enzymes accumulate genome-wide DNA damage, including unrepaired double-strand breaks, small deletions, and retained IESs. All four PolX proteins can process DNA ends, but PolXa and PolXb are specifically produced during programmed genome rearrangement and possess a unique internal linker region that enhances their nuclear localization. In contrast to PolXc, we show that PolXa concentrates in nuclear foci along with core NHEJ factors and a Dicer-like enzyme involved in producing IES-specific small RNAs that regulate IES excision. We propose that these foci are the sites where excised IESs are ligated into concatemers, which serve as templates for small RNA production, linking DNA repair to RNA-mediated regulation of genome dynamics. More information: https://academic.oup.com/nar/article/53/7/gkaf286/8113560 Contact: Mireille Bétermier mireille.betermier@i2bc-paris-saclay.fr, Julien Bischerour julien.bischerour@i2bc.paris-saclay.fr
|
|
Scooped by
I2BC Paris-Saclay
April 11, 2025 5:59 AM
|
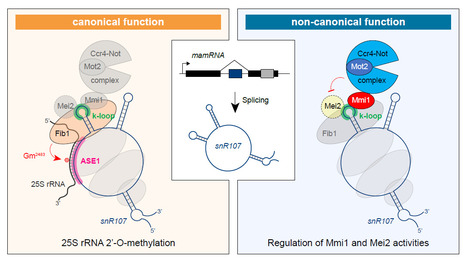
A bifunctional snoRNA guides rRNA 2’-O-methylation and scaffolds gametogenesis effectors
This study uncovers a fission yeast small nucleolar RNA (snoRNA) that guides ribosomal RNA 2’- O-methylation and modulates the activities of RNA-binding proteins involved in gametogenesis, expanding our vision of the non-canonical functions exerted by snoRNAs. Small nucleolar RNAs are non-coding transcripts that guide chemical modifications of RNA substrates and modulate gene expression at the epigenetic and post-transcriptional levels. However, the extent of their regulatory potential and the underlying molecular mechanisms remain poorly understood. In a collaborative work with the I2BC B3S department and NGS facility, the epiRNA-Seq facility in Nancy and the Palancade lab at the Institut Jacques Monod, we have identified a conserved, previously unannotated intronic C/D-box snoRNA, termed snR107, hosted in the fission yeast long non-coding RNA mamRNA and carrying two independent cellular functions. On the one hand, snR107 guides site-specific 25S rRNA 2’-O-methylation and promotes pre-rRNA processing and 60S subunit biogenesis. On the other hand, snR107 associates with the gametogenic RNA-binding proteins Mmi1 and Mei2, mediating their reciprocal inhibition and restricting meiotic gene expression during sexual differentiation. Both functions require distinct cis-motifs within snR107, including a conserved 2’-O-methylation guiding sequence. Together, our results position snR107 as a dual regulator of rRNA modification and gametogenesis effectors, expanding our vision on the non-canonical functions exerted by snoRNAs in cell fate decisions. More Information: https://www.nature.com/articles/s41467-025-58664-y Contact: Mathieu Rougemaille mathieu.rougemaille@i2bc.paris-saclay.fr
|
|
Scooped by
I2BC Paris-Saclay
March 11, 2025 5:05 PM
|
Borders in our chromosomes shape the neighboring domains
Researchers at the I2BC and the Ecole Normale Supérieure found that the borders that create separation between functional domains in our chromosomes have an unexpectedly large influence on those domains themselves. Chromosomes in humans and other mammals are subdivided into “functional neighborhoods” that guide the fidelity of biological processes, including gene regulation and DNA repair. Perturbations of these neighborhoods, resulting in the fusion of adjacent domains, can lead to a variety of diseases, including cancer and developmental disorders.
In a study published in PNAS (Proceedings of the National Academy of Sciences of the USA), scientists from the Noordermeer group at the I2BC, together with their colleagues at the Ecole Normale Supérieure in Paris, report how chromosomal border elements influence the organization of the adjacent functional neighborhoods. By combining genomics-based analysis and biophysical simulations of chromosome structure, they find that the size of these borders is highly diverse, from simple “points” on the chromosome to highly extended zones of transition between neighborhoods. Computer simulations of chromosome behavior of different types of borders in-between revealed an unexpected and far-reaching impact of the borders on their neighboring domains. Rather than creating a static separation, the borders will actively influence the degree of neighborhood mixing due to a previously unrecognized mechanism whereby the borders reel-in the adjacent neighborhoods. At narrow borders, this actively promotes mixing and may cause interference between biological activity on both side of the border. Extended borders buffer against this mechanism by adding additional separation. Border structure can thus constitute a new regulatory layer in the genome to fine-tune biological processes. More information: https://www.pnas.org/doi/10.1073/pnas.2413112122 Contact: Daan Noordermeer daan.noordermeer@i2bc.paris-saclay.fr
|
|
Scooped by
I2BC Paris-Saclay
March 4, 2025 2:57 AM
|
Ribosome profiling and immunopeptidomics reveal tens of novel conserved HIV-1 ORF encoding T cell antigens.
The translatome of HIV-1 reveals tens of alternative open reading frames (ARF), encoding conserved viral antigens. In vivo, ARF-derived peptides elicit potent HIV-specific poly-functional T cell responses mediated by both CD4+ and CD8+ T cells. T lymphocytes play a pivotal role in controlling human immunodeficiency virus type 1 (HIV-1) infection. Their activation relies on the recognition of viral peptides, or antigens, presented on the surface of infected cells by major histocompatibility complex (MHC) molecules. It is widely assumed that these antigens arise solely from canonical HIV proteins. Previously, we showed that HIV-specific T lymphocytes can also target peptides derived from HIV alternative reading frames (ARFs) presented by MHC molecules. However, evidence for ARFs in the HIV genome had thus far been indirect. In this new study, using ribosome profiling (RiboSeq), we identified the complete HIV-1 translatome in infected CD4⁺ T cells. This approach systematically identified virally encoded mRNA sequences actively translated in HIV-infected cells. We found that the HIV-1 genome contains over one hundred ARFs located either in the 5ʹ untranslated region (5ʹUTR) of classical viral genes or overlapping canonical HIV open reading frames. Using two complementary methods: (1) detecting T lymphocytes specific to ARF-derived peptides in PBMCs from people living with HIV and (2) directly isolating ARF-derived peptides bound to MHC molecules via mass spectrometry–based immunopeptidomics—we demonstrate that HIV ARFs encode novel viral antigens capable of eliciting broad and potent T-cell responses. Our results expand the range of HIV antigens that could be harnessed for vaccine development and may also reveal the existence of microproteins or pseudogenes in the HIV genome.
These findings stem from the work of two complementary teams at I2BC: one led by Olivier Namy, a specialist in protein translation, and the other led by Arnaud Moris, an expert in the immune response to HIV. This collaboration also involved French research and clinical teams and the University of Tübingen in Germany. More information: https://doi.org/10.1038/s41467-025-56773-2 Contact: Arnaud Moris arnaud.moris@i2bc.paris-saclay.fr & Olivier Namy olivier.namy@i2bc.paris-saclay.fr Image: Sonhita Chakraborty
|
|
Scooped by
I2BC Paris-Saclay
January 17, 2025 4:17 AM
|
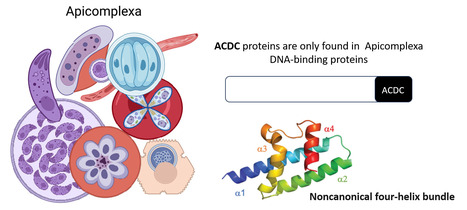
Rocking Science: New ACDC Protein Fold Unveiled
The Apicomplexa-specific ACDC domain adopts a never before seen protein fold. In collaboration with the Nessler team at the I2BC and the Llinás lab at PSU, US, we reveal that the ACDC domain, only found in DNA-binding proteins of the Apicomplexa phylum, grouping several important human pathogens including malaria, has a never before seen protein fold. We also identify potential ligands that may be optimized in the future as protein inhibitors. More information:https://journals.iucr.org/paper?S2059798324012518 Conatct: Joana SANTOS joana.santos@i2bc.paris-saclay.fr
|
|
Scooped by
I2BC Paris-Saclay
December 9, 2024 3:44 AM
|
Ribosome composition plays a key role in EMT
Ribosome remodeling drives cancer cell transformation: A single protein, RPL36A, triggers EMT. Epithelial-mesenchymal transition (EMT) is a biological process whereby a cell loses its usual characteristics to acquire properties allowing it to move. EMT is involved in embryo formation and tissue repair in adults. However, when altered, EMT contributes to the formation of fibrosis or, in the case of a tumor cell, to the formation of metastases. In cancers, this change is comparable to a cellular metamorphosis that allows cancer cells to become more aggressive and resistant to treatments. EMT is therefore a key player in tumor progression.
EMT is extensively studied at the molecular and cellular levels because a better understanding of this process will enable the development of new therapeutic approaches. However, the translational changes that occur during EMT have so far been very poorly studied.
In this work, we have identified that the overexpression of a single ribosomal protein, called RPL36A, is sufficient to induce EMT. We have also highlighted the various translational changes that occur during EMT.
Our work reveals the importance of ribosome remodeling itself in finely modulating translation during the change in cellular identity that occurs during EMT. More information : https://www.pnas.org/doi/10.1073/pnas.2408114121 ; In French: https://www.insb.cnrs.fr/fr/cnrsinfo/des-modifications-de-la-composition-du-ribosome-accompagnent-les-changements. Contact: Olivier NAMY olivier.namy@i2bc.paris-saclay.fr
|
|
Scooped by
I2BC Paris-Saclay
November 19, 2024 3:27 AM
|
The ribosome profiling landscape of yeast
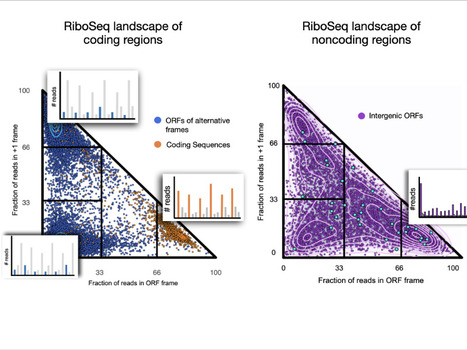
We explored the ribosome profiling landscape of yeast and showed that noncoding regions are associated with a wide diversity of translation signals among which some led to detectable protein products. Pervasive translation is a widespread phenomenon that plays a critical
role in the emergence of novel microproteins, but the diversity of translation patterns contributing to their generation remains unclear. Based on 54 ribosome profiling (Ribo-Seq) datasets, we investigated the yeast Ribo-Seq landscape using a representation framework that allows the comprehensive inventory and classification of the entire diversity of Ribo-Seq signals, including non-canonical ones. We show that if coding regions occupy specific areas of the Ribo-Seq landscape, noncoding regions encompass a wide diversity of Ribo-Seq signals and, conversely, populate the entire landscape. Our results show that pervasive translation can, nevertheless, be associated with high specificity, with 1055 noncoding ORFs exhibiting canonical Ribo-Seq signals. Using mass spectrometry under standard conditions or proteasome inhibition with an in-house analysis protocol, we report 239 microproteins originating from noncoding ORFs that display canonical but also non-canonical Ribo-Seq signals. Each condition yields dozens of additional microprotein candidates with comparable translation properties, suggesting a larger population of volatile microproteins that are challenging to detect. Our findings suggest that non-canonical translation signals may harbor valuable information and underscore the significance of considering them in proteogenomic studies. Finally, we show that the translation outcome of a noncoding ORF is primarily determined by the initiating codon and the codon distribution in its two alternative frames, rather than features indicative of functionality. Our results enable us to propose a topology of a species’ Ribo-Seq landscape, opening the way to comparative analyses of this translation landscape under different conditions. More information: https://genomebiology.biomedcentral.com/articles/10.1186/s13059-024-03403-7 Contact: Anne Lopes anne.lopes@i2bc.paris-saclay.fr
|
|
Scooped by
I2BC Paris-Saclay
November 3, 2024 12:00 PM
|
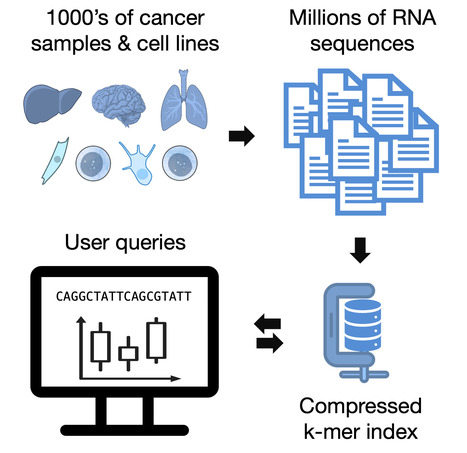
Reference-free exploration of cancer RNA datasets
Bioinformaticians finds new ways to analyze cancer RNA in very large datasets Cancer cells produce an immense diversity of RNA molecules. Using deep sequencing technologies, scientists explore these RNAs to uncover key insights into cancer onset and progression. However, cancer RNA datasets are becoming so large that directly searching them for a given sequence is impossible. Currently, scientists must rely on simplified representations, such as gene expression tables. Yet, many cancer-causing RNAs are absent from standard gene expression tables. The Transipedia consortium, bringing together bioinformaticians from I2BC/University Paris-Saclay, University of Lille, and University of Montpellier, introduces a new approach to mining RNA-seq databases using k-mer indexes. In their latest paper, they demonstrate that an index of 1,019 cancer cell lines can be queried in real time while providing accurate quantification of cancer mutations, fusions, or splicing variants. In future work, the consortium plans to expand this technology to offer access to an even larger database of cancer and normal tissues, aiming to accelerate cancer genomics research. More information: https://genomebiology.biomedcentral.com/articles/10.1186/s13059-024-03413-5 Contact: Daniel Gautheret daniel.gautheret@i2bc.paris-saclay.fr
|
|
Scooped by
I2BC Paris-Saclay
October 29, 2024 5:36 AM
|
Novel, tightly structurally related N-myristoyltransferase inhibitors display equally potent yet distinct inhibitory mechanisms
Peptides fitting the optimal human NMT Gly-myristoylation recognition pattern act as potent inhibitors. Lys-myristoylation-based inhibitors from these peptides were designed. Each series’ inhibitory properties are unique, relying on distinct interactions. N-myristoyltransferases (NMTs) catalyze essential acylations of N-terminal alpha or epsilon amino groups of glycines or lysines. Here, we reveal that peptides tightly fitting the optimal glycine recognition pattern of human NMTs are potent prodrugs relying on a single-turnover mechanism. Sequence scanning of the inhibitory potency of the series closely reflects NMT glycine substrate specificity rules, with the lead inhibitor blocking myristoylation by NMTs of various species. We further redesigned the series based on the recently recognized lysine-myristoylation mechanism by taking advantage of (i) the optimal peptide chassis and (ii) lysine side chain mimicry with unnatural enantiomers. Unlike the lead series, the inhibitory properties of the new compounds rely on the protonated state of the side chain amine, which stabilizes a salt bridge with the catalytic base at the active site. Our study provides the basis for designing first-in-class NMT inhibitors tailored for infectious diseases and alternative active site targeting. More information: https://www-cell-com.insb.bib.cnrs.fr/structure/abstract/S0969-2126(24)00318-6?_returnURL=https%3A%2F%2Flinkinghub.elsevier.com%2Fretrieve%2Fpii%2FS0969212624003186%3Fshowall%3Dtrue Contact: Thierry Meinnel thierry.meinnel@i2bc.paris-saclay.fr
|
|
|
Scooped by
I2BC Paris-Saclay
January 13, 3:52 PM
|
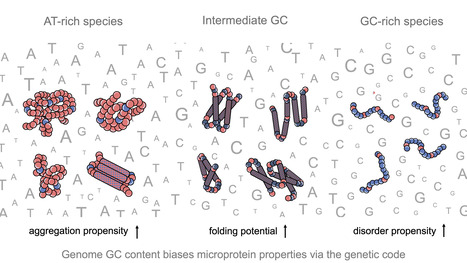
Impact of GC content on de novo gene birth
Noncoding DNA can generate microproteins, some of which evolve into new genes. We show that de novo genes preferentially originate from GC-rich, foldable sequences, revealing how base composition channels the birth of new proteins. Noncoding regions of eukaryotic genomes are widely transcribed and constitute a major source of novel microproteins, some of which eventually become fixed as de novo genes - a process known as de novo gene birth that plays a significant role in species adaptation. However, the structural properties of these nascent proteins and the factors governing their evolutionary fate remain poorly understood. In particular, the role of genome nucleotide composition (GC content) in shaping their biophysical properties has remained unclear. In this study, researchers of the I2BC, analyzed the foldability and sequence properties of millions of putative microproteins encoded by intergenic open reading frames (ORFs) from 3,379 eukaryotic species spanning a broad range of GC contents (18–79%). Results show that GC content strongly influences amino-acid composition and structural tendencies, suggesting distinct cellular impacts if non-genic regions are pervasively expressed. AT-rich species predominantly encode ORFs biased toward hydrophobic, aggregation-prone sequences, whereas GC-rich species tend to encode more hydrophilic, disorder-prone ORFs. ORFs from genomes with intermediate GC content display a more balanced composition and higher folding potential, with many expected to adopt proto-folds. To assess how these properties relate to gene emergence,the authors traced the evolutionary history of several hundred de novo proteins across 22 species using phylostratigraphy, targeted de novo gene searches, and ancestral sequence reconstruction. Researchers find that de novo genes preferentially originate from GC-rich ORFs with intrinsic folding potential. Together, our results reveal that the interplay between GC content and foldability - rooted in the structure of the genetic code - shapes the emergence of novel genes. More information: https://www.nature.com/articles/s41467-025-68022-7 Contact: Anne Lopes anne.lopes@i2bc.paris-saclay.fr
|
|
Scooped by
I2BC Paris-Saclay
January 6, 4:23 PM
|
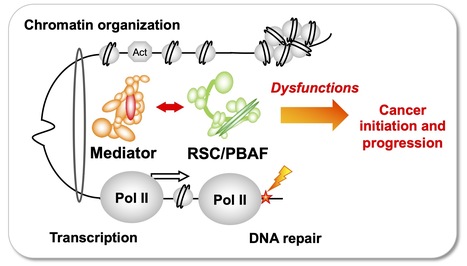
Team of J. Soutourina received the label from Ligue Nationale contre le cancer
Understanding how the essential complexes —the coregulator Mediator and the chromatin remodeling complex RSC/PBAF— cooperate in the nucleus In eukaryotes, transcription and DNA repair occur in the crowded context of chromatin. Dysfunctions of these processes can lead to cancers. Mediator is an essential and conserved multisubunit coactivator complex, mutated in many cancers. However, it remains largely unknown how Mediator and chromatin regulators coordinate their functions. A recent publication of the team suggests the novel hypothesis that Mediator acts in conjunction with the chromatin remodeling complex RSC (Remodels the Structure of Chromatin) of SWI/SNF family, homologous to PBAF (Polybromo-associated BAF) in human, representing the most frequently mutated complexes in cancers. Building on this recent publication, a new project has been supported by Ligue Nationale contre le cancer. Using the yeast model, with a perspective to extend the study to human cells, the team intends to decipher the molecular mechanisms involved in functional cooperation between these essential coregulator complexes in transcription regulation, DNA repair and chromatin organization relevant for cancer biology. Contact: Julie Soutourina julie.soutourina@i2bc.paris-saclay.fr
|
|
Scooped by
I2BC Paris-Saclay
November 9, 2025 4:21 AM
|
Three stop codons to get over to flourish
Funded by the European Research Council (ERC synergy 2025), the project “3Stops2Go” leaps over the “three red lights” of premature stop codons to re-express critical protein and correct genetic diseases. The project, recently funded by an ERC Synergy Grant 2025, aims to target premature termination codon (PTC)mutations, which prematurely halt protein translation and are involved in about 11% of human genetic diseases.
By combining expertise in protist biology, RNA biology, and gene therapy, the consortium of four researchers (Leoš Valášek (Institute of Microbiology, Czech Academy of Sciences, Czech Republic), Julius Lukeš (Biology Centre, Czech Academy of Sciences, Czech Republic), Olivier Namy (Université Paris-Saclay, CEA, CNRS, France / I2BC, France), and Mark Osborn (University of Minnesota, USA)) aims to harness natural stop-codon bypass mechanisms to develop therapeutic tools (engineered tRNAs and readthrough inducers), test them in patient-derived cells and animal models, and ultimately pave the way toward clinical applications. More information: https://erc.europa.eu/news-events/news/synergy-grants-2025-examples-projects Contact: Olivier Namy olivier.namy@i2bc.paris-saclay.fr>
|
|
Scooped by
I2BC Paris-Saclay
April 14, 2025 3:19 AM
|
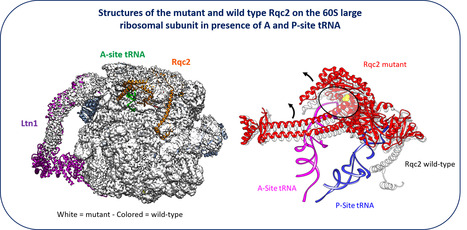
New facet of the Rqc2 protein part of a ribosome rescue pathway in Saccharomyces cerevisiae
The Ribosome Quality Complex protein 2 is a major player in peptide release from stalled ribosomes. Cells employ many quality control mechanisms during protein biosynthesis. Defects impairing messenger RNA decoding can cause ribosomes to stall, thereby distorting gene expression. Ribosome stalling results in ribosomes being sequestered in an inactive state on mRNA and represents a challenge for both ribosome homeostasis and cell survival. Eukaryotic cells prevent the accumulation of potentially toxic aberrant polypeptides and maintain ribosome availability by employing surveillance and clearance mechanisms, including the evolutionarily conserved ribosome-associated quality control complex (RQC). The rescue pathways dissociate the stalled ribosome, leading to the release of a 40S subunit attached to the mRNA and a 60S subunit still attached to a tRNA with the elongating peptide. RQC mediates the ubiquitination, extraction, and rapid degradation of the aberrant nascent peptide. In particular, Rqc2p recruits charged alanyl- and threonyl-tRNAs at the same position as an A-site tRNA, resulting in C-terminal extensions composed of alanine and threonine residues (CAT tails). These CAT tails may expose potential tunnel-embedded lysine residues for ubiquitination by the E3 ligase Ltn1. Using a genetic screen, we identified a mutant allele of RQC2 as involved in peptide release from stalled ribosomes. We characterized the mutant protein both biochemically and structurally, in collaboration with Reynald Gillet’s team (IGDR, Rennes University). We determining how the underlying point mutation reduced tRNA recruitment to the A site, thereby limiting CAT tail formation. These findings provide further mechanistic insight into the role of Rqc2p in ribosome rescue and the maintenance of ribosome homeostasis. More Information:https://pubmed.ncbi.nlm.nih.gov/40187343/ Contact: Céline Fabret celine.fabret@i2bc.paris-saclay.fr
|
|
Scooped by
I2BC Paris-Saclay
April 7, 2025 5:04 AM
|
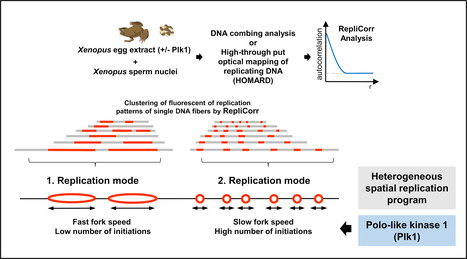
Yin-Yang during DNA replication
DNA duplication of the genetic material is a crucial step of cell proliferation. The study reveals that this step comprises two opposed strategies, coordinated by the enzyme Plk1. In vertebrates, DNA replication involves the activation of thousands of starting points for DNA copying, known as “origins of replication”. These points are irregularly scattered across the genome. However, the precise mechanisms controlling the coordinated activation of these starting points and the regulation of the rate at which DNA is copied remain poorly understood. Any dysfunction can lead to genetic abnormalities and promote the development of cancers.
The scientists studied DNA replication using cell-free extracts from the eggs of the amphibian Xenopus laevis. This model is quite similar to replication in human cells, and has made it possible to develop a new method for analyzing the distribution of replication starting points (initiations) and the speed of replication in individualized DNA molecules. Combining this analysis with kinetic models, the scientists discovered that DNA replication relies on two dynamic and opposing strategies:
A fast strategy: high replication speed, but with fewer starting points.
A slow strategy: a slower replication speed compensated by the activation of numerous starting points.
In this context, the scientists determined that the enzyme Polo-like kinase 1 (Plk1) played a key role in coordinating these two strategies. Plk1, frequently mutated in many cancers, regulates this balance, opening up new perspectives on the control of replication and its implications in oncology. (published by Paris-Saclay and CNRS Biologie Actu (https://www.insb.cnrs.fr/fr/cnrsinfo/yin-yang-dans-la-duplication-des-chromosomes ) More Information: https://doi.org/10.1093/nar/gkaf007 Contact: Arach Goldar arach.goldar@i2bc.paris-saclay.fr Kathrin Marheineke kathrin.marheineke@ijm.fr
|
|
Scooped by
I2BC Paris-Saclay
March 4, 2025 3:20 AM
|
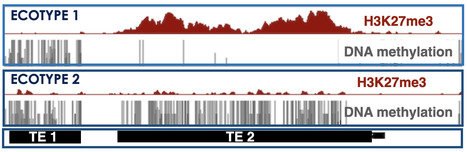
Alternative silencing states of transposable elements in Arabidopsis associated with H3K27me3
This study sheds light on an alternative mode of Transposable Element silencing associated with H3K27me3 instead of DNA methylation in flowering plants; it also indicates dynamic switching between the two epigenetic marks at the species level, a new paradigm that might extend to other multicellular eukaryotes. The DNA/H3K9 methylation and Polycomb-group proteins (PcG)-H3K27me3 silencing pathways have long been considered mutually exclusive and specific to transposable elements (TEs) and genes, respectively in mammals, plants, and fungi. However, H3K27me3 can be recruited to many TEs in the absence of DNA/H3K9 methylation machinery and sometimes also co-occur with DNA methylation.
In this study, the team of Angélique Déléris showed that TEs can also be solely targeted and silenced by H3K27me3 in wild-type Arabidopsis plants. These H3K27me3-marked TEs not only comprise degenerated relics but also seemingly intact copies that display the epigenetic features of responsive PcG target genes as well as an active H3K27me3 regulation. The authors also showed that H3K27me3 can be deposited on newly inserted transgenic TE sequences in a TE-specific manner indicating that silencing is determined in cis. Finally, a comparison of Arabidopsis natural accessions reveals the existence of a category of TEs—which we refer to as “bifrons”—that are marked by DNA methylation or H3K27me3 depending on the accession. This variation can be linked to intrinsic TE features and to trans-acting factors and reveals a change in epigenetic status across the TE lifespan.
Overall, this study shedded light on an alternative mode of TE silencing associated with H3K27me3 instead of DNA methylation in flowering plants. It also suggests dynamic switching between the two epigenetic marks at the species level, a new paradigm that might extend to other multicellular eukaryotes. More Information: https://genomebiology.biomedcentral.com/articles/10.1186/s13059-024-03466-6 Contact: Angélique Déléris angelique.deleris@i2bc.apris-saclay.fr
|
|
Scooped by
I2BC Paris-Saclay
January 30, 2025 3:50 AM
|
Genetic differentiation in the MAT-proximal region is not sufficient for suppressing recombination in Podospora anserina
A large genomic region in Podospora anserina remains entirely devoid of crossovers, despite being fully colinear. Recombination is advantageous over the long-term, as it allows efficient selection and purging deleterious mutations. Nevertheless, recombination suppression has repeatedly evolved in sex chromosomes. In fungi, sexual compatibility is driven by a locus called mating-type (mat), around which recombination suppression is also often observed.
The evolutionary causes for recombination suppression and the proximal mechanisms preventing crossing overs are poorly understood. Several hypotheses have recently been suggested based on theoretical models, and in particular, that divergence could accumulate neutrally around a sex-determining region and reduce recombination rates, a self-reinforcing process that could foster progressive extension of recombination suppression.
We used the ascomycete fungus Podospora anserina for investigating these questions: a 0.8 Mbp region around its mat locus is non-recombining (called MAT-proximal region), despite being collinear between the two mating types. This fungus is mostly selfing, resulting in highly homozygous individuals, except in the non-recombining region around the mating-type locus that displays differentiation between mating types. Here, we test the hypothesis that sequence divergence alone is responsible for recombination cessation. We replaced one mat idiomorph by the sequence of the other, to obtain compatible strains isogenic in the MAT-proximal region. Crosses showed that recombination was still suppressed in that context, indicating that other proximal mechanisms than inversions or mere sequence divergence are responsible for recombination suppression in this fungus.
This finding suggests that selective mechanisms likely acted for suppressing recombination, or the spread of epigenetic marks, as the neutral model based on mere nucleotide divergence does not seem to hold in P. anserina. More information: https://doi.org/10.1093/g3journal/jkaf015 Contact: Pierre GROGNET pierre.grognet@universite-paris-saclay.fr
|
|
Scooped by
I2BC Paris-Saclay
December 16, 2024 5:49 AM
|
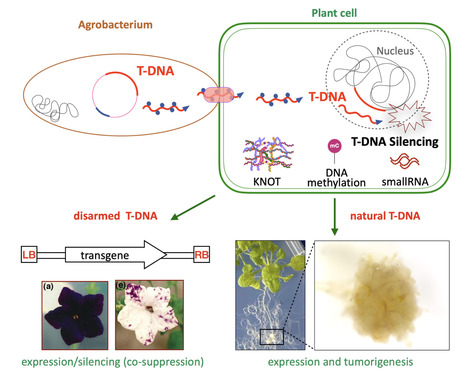
Epigenetic control of T-DNA during transgenesis and pathogenesis
T-DNAs are mobile elements transferred from pathogenic Agrobacterium to plants that reprogram host cells into hairy roots or tumors, and which are used as disarmed forms to deliver transgenes in plants. Here we review the mechanisms that silence the expression of T-DNAs in transgenic plants as well as during pathogenesis. Mobile elements known as T-DNAs are transferred from pathogenic Agrobacterium to plants and reprogram the host cell to form hairy roots or tumors. Disarmed nononcogenic T-DNAs are extensively used to deliver transgenes in plant genetic engineering. Such T-DNAs were the first known targets of RNA silencing mechanisms, which detect foreign RNA in plant cells and produce small RNAs that induce transcript degradation. These T-DNAs can also be transcriptionally silenced by the deposition of epigenetic marks such as DNA methylation and the dimethylation of lysine 9 (H3K9me2) in plants. Here, we review the targeting and the roles of RNA silencing and DNA methylation on T-DNAs in transgenic plants as well as during pathogenesis. In addition, we discuss the crosstalk between T-DNAs and genome-wide changes in DNA methylation during pathogenesis. We also cover recently discovered regulatory phenomena, such as T-DNA suppression and RNA silencing-independent and epigenetic-independent mechanisms that can silence T-DNAs. Finally, we discuss the implications of findings on T-DNA silencing for the improvement of plant genetic engineering. More information: https://academic-oup-com.insb.bib.cnrs.fr/plphys/advance-article/doi/10.1093/plphys/kiae583/7876130 Contact: Angélique DELERIS angelique.deleris@i2bc.paris-saclay.fr
|
|
Scooped by
I2BC Paris-Saclay
November 19, 2024 3:50 AM
|
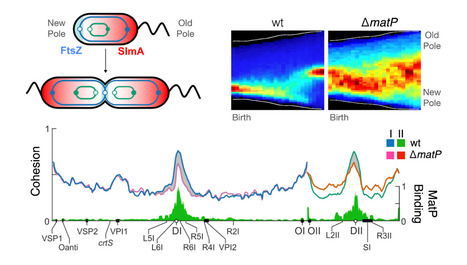
MatP local enrichment delays segregation independently of tetramer formation and septal anchoring in Vibrio cholerae
The study of Vibrio cholerae MatP reveals the degree of integration of the plasmid-derived chromosome carried by the bacterium, and suggests that the ancestral role of MatP is to promote cell division at the end of the chromosome replication and segregation cycle. V. cholerae harbours a primary chromosome derived from the monochromosomal ancestor of the Vibrionales (ChrI) and a secondary chromosome derived from a megaplasmid (ChrII). The coordinated segregation of the replication terminus of both chromosomes (TerI and TerII) determines when and where cell division occurs. ChrI encodes a homolog of Escherichia coli MatP, a protein that binds to a DNA motif (matS) that is overrepresented in replication termini. Here, we use a combination of deep sequencing and fluorescence microscopy techniques to show that V. cholerae MatP structures TerI and TerII into macrodomains, targets them to mid-cell during replication, and delays their segregation, thus supporting that ChrII behaves as a bona fide chromosome. We further show that the extent of the segregation delay mediated by MatP depends on the number and local density of matS sites, and is independent of its assembly into tetramers and any interaction with the divisome, in contrast to what has been previously observed in E. coli. More information: https://www.nature.com/articles/s41467-024-54195-0 Contact: E. Galli elisa.galli@i2bc.paris-saclay.fr & FX Barre francois-xavier.barre@i2bc.paris-saclay.fr
- Are you interested in Parasitology? or - Would you like to discover a field of research at the interface between Biology and Chemistry? or - Would you like to discover new research methods or study models? Come and attend the Workshop: Practical information: - Date: 11/27/2024
- Times: 9.15 a.m. to 12.15 p.m. (detailed program above)
- Address: Bâtiment Henri Moissan, 17, avenue des Sciences, 91400 ORSAY, France
- Room: 2000 (HM1 building)
- Program and theme (details attached): 3 parasitology specialists come to talk about their latest research findings on malaria and leishmaniasis. Come and listen to their lectures and have lunch with them.
- Registration (deadline 11/20/2024): registration is free but compulsory by clicking on this link
- Module validation: This seminar could validate a doctoral school module for doctoral students.
Via Life Sciences UPSaclay
|
|
Scooped by
I2BC Paris-Saclay
October 30, 2024 8:53 AM
|
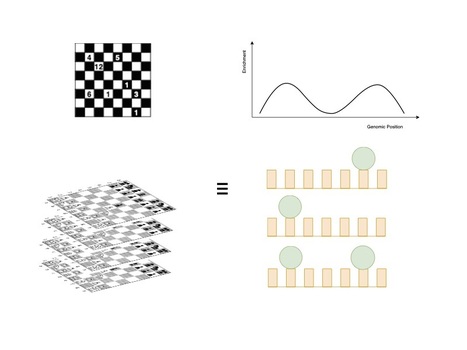
The next-generation sequencing—chess problem
By comparing the collection of Next-Generation Sequencing data to the superimposition of several chess games, we demonstrate the limits of current temporal analysis approaches. The development of next-generation sequencing (NGS) technologies paved the way for studying the spatiotemporal coordination of cellular processes along the genome. However, data sets are commonly limited to a few time points, and missing information needs to be interpolated. Most models assume that the studied dynamics are similar between individual cells, so that a homogeneous cell culture can be represented by a population-wide average. Here, we demonstrate that this understanding can be inappropriate. We developed a thought experiment—which we call the NGS chess problem—in which we compare the temporal sequencing data analysis to observing a superimposed picture of many independent games of chess at a time. The analysis of the spatiotemporal kinetics advocates for a new methodology that considers DNA-particle interactions in each cell independently even for a homogeneous cell population. More information: https://academic.oup.com/nargab/article/6/4/lqae144/7833696 Contact: Julie Soutourina julie.soutourina@i2bc.paris-saclay.fr
|