Your new post is loading...
Your new post is loading...

|
Scooped by
Gilbert C FAURE
November 9, 2022 1:39 AM
|
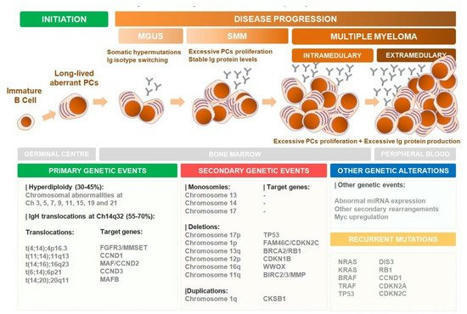
Cell-specific DTA expression depletes BMAds. We recently created a potentially novel BMAd-specific Cre mouse model (6) that uses an intersectional strategy with dual recombinases: Osterix-FLPo and codon-optimized flippase recombinase (FLPo)-dependent Adipoq-Cre (FAC). Briefly, endogenous Osterix-driven FLPo recognizes Frt sites and flips the reverse orientation of the Cre cassette located in the 3′-untranslated region of endogenous Adipoq into the sense orientation (Supplemental Figure 1, A–C; supplemental material available online with this article; https://doi.org/10.1172/jci.insight.160915DS1). Osterix traces to osteoblasts and BMAds (18–20), and adiponectin is expressed in all mature adipocytes, including BMAds (21). Thus, the Osterix-FLPo–activated Adipoq-Cre recombinase is predominantly expressed in the overlapping BMAd population. To confirm cell specificity and efficiency of this BMAd-Cre mouse model, mT/mG reporter mice (22) were used to visualize Cre activity. EGFP+ cells were observed in BMAd, but not in bone cells, epididymal white adipose tissue (eWAT), or other tissues/organs evaluated (Supplemental Figure 1, D and E) (6). Of note, efficiency of BMAd-Cre increases with age, and recombination is less than 90% when mice have 2 copies of FAC and are younger than 12 weeks of age (6). There is increasing evidence for BMAds having a specific marrow adipogenic lineage precursoe (MALP) that expresses Adipoq, Cxcl12, and Lepr (23, 24). These pericytic and stromal cells have a central cell body and have dendritic processes that extend through the marrow niche. MALPs are traced by constitutive and inducible Adipoq-based Cre and compose up to 0.6% of marrow cellularity (24). BMAd-Cre mice appear to target only a small subset of MALPs (Supplemental Figure 1, F–H) (6), perhaps because many are not lineage traced by Osterix or perhaps because expression of Cre from the endogenous Adipoq locus is less within this cell population relative to BMAds. To investigate physiological functions of BMAds, we next bred BMAd-Cre mice to ROSA-DTA mice harboring a LoxP-flanked STOP cassette proximal to the DTA sequence (Supplemental Figure 1I). In this model, Cre recombinase excises the STOP cassette to allow expression of cytotoxic DTA in BMAds, thus inducing cell death. We measured BM adiposity in osmium tetroxide–stained decalcified tibiae from these mice and observed a marked reduction of rBMAT in proximal tibiae and cBMAT in distal tibiae (Figure 1A); these data were further confirmed by histological analyses of H&E-stained bone sections (Figure 1B) and quantification of osmium tetroxide by μCT (Figure 1C). Compared with control littermates, BMAd-DTA mice did not have altered total body weights, random glucose concentrations, or soft tissue weights (Supplemental Figure 2, A–C). Histological analysis of s.c. WAT (sWAT) and eWAT was comparable between BMAd-DTA and control mice (Supplemental Figure 2, D and E). Bulk RNA-Seq analysis of distal tibiae revealed that both adipogenesis and fatty acid metabolism pathways were downregulated in BMAd-DTA mice compared with controls (Figure 1D), consistent with observed reduction in BMAT content. These data were confirmed by quantitative PCR (qPCR) analysis showing that Pparg and Cebpa expression in distal tibiae are decreased in BMAd-DTA mice (Supplemental Figure 2F). We next isolated BM mesenchymal stem cells (BMSCs) and induced adipogenic differentiation. Cultures of DTA BMSCs did not have detectable lipid-containing adipocytes 21 days after induction of adipogenesis (Figure 1E). Consistent with DTA causing adipocyte cytotoxicity, adipocyte genes such as Adipoq and Fabp4 were nearly undetectable in these cells (Figure 1F). Figure 1DTA expression depletes BMAds. Control (–) and BMAd-DTA (+) male mice at 20–24 weeks of age were sacrificed to validate the depletion of BMAds by DTA expression (n = 7 per group). Experiments were repeated more than 3 times. (A) Tibiae were decalcified with 14% EDTA for 3 weeks and stained with 1% osmium tetroxide for 48 hours. μCT scanning was performed, and 3D reconstituted images of tibiae are shown. (B) Decalcified bones were processed for paraffin embedding and sectioning. Paraffin slides were stained with H&E. Images were taken under 100× magnification. Scale bars: 200 μm. (C) Lipid staining within the BM was quantified by μCT scanning following osmium tetroxide staining. Proximal metaphysis is the region indicated by 1 to 2, and distal tibia is the region between 3 and 4, shown in A. Data are presented as mean ± SD. *P < 0.05 with a 2-tailed t test. (D) Fresh distal tibiae were collected and hammered into powder for bulk RNA purification. RNA-Seq and GSEA were performed. (E and F) BM mesenchymal stem cells were isolated from control and BMAd-DTA male mice at 16 weeks of age and were differentiated into adipocytes. Scale bar: 200 μm. After 3 weeks of differentiation, cells were collected for RNA purification, followed by qPCR. Fold changes of gene expression were normalized to the geomean of Hprt and Rpl32a. Data are presented as mean ± SD. *P < 0.05 with 2-way ANOVA with Šídák’s multiple-comparison test. Loss of BMAds depletes hematopoietic stem and progenitor cells (HSPCs). To date, studies exploring the potential role of BMAds in hematopoiesis have yielded conflicting conclusions — that BMAds impair hematopoietic regeneration or that BMAds promote hematopoiesis by secreting stem cell factors (7, 25). Our prior findings support a positive role for BMAds in hematopoieses that BMAd lipolysis is required for myelopoiesis in CR or irradiated mice (6). To determine effects of removing majority of BMAd-derived factors, we collected and analyzed whole blood from controls and BMAd-DTA mice and did not observe significant changes in circulating WBC or RBC populations. Flow cytometry analysis of hematopoietic cells isolated from long bones revealed that, although overall BM nucleated cells and T cells were decreased with loss of BMAT, most mature blood cell populations were not significantly changed (Supplemental Figure 3, A–E). In addition, HSPC populations were significantly reduced in BMAd-DTA mice, including hematopoietic stem cells (HSC), multipotent progenitors (MPP), hematopoietic progenitor cell 1 (HPC1), HPC2, premegakaryocyte/erythrocyte progenitors (PreMegE), and pre–CFU erythroid (preCFUe) precursors (Supplemental Figure 3, F–K). Furthermore, evaluation of CFU showed impaired proliferative capacity of most HSPCs isolated from tibiae of BMAd-DTA mice, including multipotential (CFU-granulocyte, erythrocyte, monocyte, megakaryocyte [GEMM]), granulocyte macrophage (CFU-GM), granulocyte (CFU-G), macrophage (CFU-M), and B lymphocyte (CFU-preB) progenitor cells, but it did not show impaired proliferative capacity of erythroid cell (CFU-E) (Supplemental Figure 3, L–Q). Interestingly, CFU-GEMM sizes were significantly smaller in BMAd-DTA mice compared with controls (Supplemental Figure 3, R and S). In support for this observation, RNA-Seq data reveal that pathways related to cell cycle, cell division, cell cycle checkpoints, DNA, and replication were extensively suppressed in BMAd-DTA mice (Supplemental Figure 3T). To investigate the mechanisms underlying alterations in these cell populations, we performed qPCR analysis on RNA isolated from sorted HSCs and MPPs and found that c-Myc gene expression was suppressed in MPPs derived from BMAd-DTA mice (Supplemental Figure 3, U and V). Although MYC is one of the master regulators required for hematopoietic cell growth, future studies are required to explore these mechanisms in more detail. BMAT depletion is accompanied by increased cortical bone formation. Given the significant changes observed within the BM niche following BMAT depletion, one might assume that development or homeostasis of bone turnover would also be affected. As mentioned above, BMAd-Cre recombinase was less efficient in BMAd-Cre mice younger than 12 weeks of age; therefore, we did not observe significant differences in length of body or long bones (Supplemental Figure 4, A–C), suggesting that bone growth remains largely unaffected; however, lumbar 5 (L5) and caudal vertebra 5 (CV5) were shorter in BMAd-DTA mice (Supplemental Figure 4D). Although loss of rBMAT in proximal tibiae was accompanied by increased trabecular bone (Tb.) volume fraction (BV/TV), mineral density (BMD), connective density (Conn. Dens), and number (N) (Supplemental Figure 4, E–G), elevated proximal tibial trabecular bone was only observed in some experimental cohorts. In contrast, higher distal tibial cortical parameters such as bone area fraction (Ct. bone area [BA]/total area [TA]) and cortical thickness (Ct. Th) were consistently found across all cohorts (Figure 2, A and B) without changes to total volume of marrow in distal tibiae (Supplemental Figure 4H). These site-specific skeletal effects may be due to the fact that there is less BMAT content in proximal tibiae than in distal tibiae. To further explore mechanisms underlying increased cortical bone mass, we performed dynamic histomorphometry analysis, which revealed significant increases in endosteal interlabel width (Ec. Ir. L. Wi) but no change in periosteum (Figure 2, C and D). Since Zou et al. previously showed that transgenic Adipoq-Cre-DTA mice developed osteosclerosis (9), we next used Raman microscopy to evaluate quality of endosteal, midcortical, and periosteal bone within distal tibiae (26) (Figure 2E). Consistent with loss of BMAT within the BM cavity, BMAd-DTA mice had a decreased lipid/mineral ratio in endosteal bone. Furthermore, the mineral/matrix ratio tended to be elevated in midcortical bone of BMAd-DTA mice, and crystallinity trended downward in endosteal and periosteal bone areas. Two-way ANOVA revealed a significant genotype effect, indicating that overall bone quality was improved in BMAd-DTA mice (Figure 2F). Of note, Raman collagen crosslinking (Xlinks) ratio was not different between genotypes. Figure 2Depletion of BMAds increases cortical bone formation. Control (–) and BMAd-DTA (+) male mice at 20–24 weeks of age received calcein injections 9 and 2 days before dissection. Tibiae were collected for μCT and dynamic histomorphometry (n = 8–10 per group). Experiments were repeated twice. (A) Representative images of distal tibiae from μCT scanning are shown. Scale bar: 500 μm. (B) Distal tibial cortical bone area fraction (BA/TA) and thickness (Th) were quantified for control and BMAd-DTA mice. (C and D) Calcein-labeled tibiae were cross-sectioned and imaged by fluorescence microscopy. Cortical bone interlabel widths (Ir.L.Wi) in endosteum (Ec.) and periosteum (Ps.) were quantified by BioQuant software. Data are presented as mean ± SD. *P < 0.05 with a 2-tailed t test (B and D). (E and F) Fresh distal tibial cortical bone was cross-sectioned and analyzed with Raman microscopy (n = 3–4). Representative spectra from distal tibiae are shown, and major peaks are labeled (E). Lipid/mineral ratio, mineral/matrix ratio, collagen crosslink (Xlinks), and bone crystallinity were calculated for endosteal (Endo-), mid-cortical (Mid-), and periosteal (Peri-) regions (F). Data are presented as mean ± SD. *P < 0.05 with 2-way ANOVA analyses followed by Šídák’s multiple-comparison test. (G and H) Total RNA was purified from distal tibiae and used for RNA-Seq analysis (n = 4 for control; n = 7 for BMAd-DTA). The upregulated gene set was analyzed by MetaScape to identify enriched terms and pathways (G). Z scores of genes related to ossification pathway are shown as heatmap (H). To further characterize the molecular signature following BMAT depletion, we performed RNA-Seq analysis on whole distal tibiae of control and BMAd-DTA mice. We identified 841 genes that were significantly changed in BMAd-DTA mice; of these, 432 genes were increased. Pathway analysis of upregulated genes revealed that pathways related to ossification, extracellular matrix organization, and osteoblast differentiation were activated (Figure 2G). For example, further analyses of genes clustered in the ossification pathway showed that Wnt signaling–related genes (e.g., Ctnnb1, Lrp4/5, and Wnt5a), osteoblast markers (e.g., Alpl, Bglap, and Bmp genes), and osteoblast-secreted collagen genes (e.g., Col1a1, Col1a2, and Col2a1) were induced in BMAd-DTA mice (Figure 2H), consistent with enhancement of bone formation and mineralization (e.g., Mepe and Phex). To gain additional insights into mechanisms by which BMAd depletion enhanced bone formation, we mapped the differentially regulated genes (Padj < 0.05) with a secretome database, MetazSecKB (27), and found that 89 genes were classified as secretory proteins. Pathway analysis of these 89 genes highlighted the induction of extracellular matrix organization and skeletal system development pathways (Supplemental Figure 4, I–K) and included factors such as Bgn (28), Dmp1 (29), Pcsk5 (30), and Sfrp2 (31). Since bulk RNA-Seq was perform on whole distal tibiae, we were not able to distinguish specific cellular sources for the secretory factors. Trabecular bone formation is enhanced in caudal vertebrae of BMAd-DTA mice. In addition to the distal tibiae, cBMAT is also enriched within caudal vertebrae (2). Interestingly, BMAd-specific DTA expression did not greatly reduce the amount of cBMAT observed within caudal vertebrae (Figure 3A). However, even with relatively minor changes in caudal cBMAT of BMAd-DTA mice, we observed increased trabecular number and larger populations of hematopoietic cells interspersed between caudal vertebral cBMAds (Figure 3B). Consistent with the notion of losing cBMAT in caudal vertebrae, expression of both Pparg and Cebpa were decreased in BMAd-DTA mice (Figure 3C), perhaps due to reduced number of cBMAd, as well as suppressed transcription factor expression per BMAd. We next performed μCT analysis, which revealed elevated Tb. BV/TV, Tb. BMD, Tb. N, and thickness (Tb. Th), and decreased trabecular separation (Tb. Sp) (Figure 3, D and E), consistent with visual observation of increased trabecular bone. Histomorphometry analysis showed an increased number of osteoblasts located on the trabecular bone surface in BMAd-DTA mice (Figure 3, F and G), whereas osteoclast number and eroded surface were not affected (Figure 3, H and I), indicating that elevated bone mass was largely due to enhanced bone formation rather than inhibited bone resorption. This conclusion is supported by expression of bone formation markers Alpl, Sp7, and Bglap, which were upregulated in BMAd-DTA mice compared with controls (Figure 3J). Figure 3Trabecular bone formation is enhanced in caudal vertebrae of BMAd-DTA mice. Control (–) and BMAd-DTA (+) male mice at 20–24 weeks of age were sacrificed, and the fourth through sixth caudal vertebrae were collected (2 cohorts were included: one cohort with n = 11–14 per group was used for μCT analyses; the other cohort with n = 13 per group was split into histology and qPCR). (A and B) Caudal vertebrae were decalcified and used for paraffin sectioning. H&E-stained slides were scanned for an overview of the fifth caudal vertebra (A). Higher magnification images were taken at 100× (B). Scale bar: 200 μm. (C) RNA from the fourth through sixth caudal vertebrae was purified and used for qPCR to measure expression of adipogenic transcriptional factors. Relative gene expression is presented after normalization to the geomean of Hprt and Rpl32a. (D and E) Caudal vertebral trabecular bone parameters were assessed by CT. Scale bar: 200 μm. Tb., Trabecular bone; BV/TV, bone volume fraction; BMD, bone mineral density; Conn. Dens, connective density; N, number; Th, thickness; Sp, separation. (F and G) H&E-stained slides were used to count osteoblast number (N. Ob) and normalized to bone surface (BS). (H and I) Paraffin-sectioned slides were used for TRAP staining and osteoclast (Oc) number (N) and surface (S) quantification. (J) Osteogenic gene expression in caudal vertebrae were evaluated by qPCR and normalized to the geomean of Hprt and Rpl32a. Data are presented as mean ± SD. *P < 0.05 with a 2-tailed t test. Multiple unpaired t tests were performed in E and J, and P values were adjusted for multiple comparisons using 2-stage step-up (Benjamini, Krieger, and Yekutieli) with FDR method. Loss of cBMAT protects mice from CR-induced cortical bone loss. To determine whether enhanced bone formation observed in BMAd-DTA mice counteracts CR-induced bone loss, we challenged cohorts of mice with 30% CR for 12 weeks. As expected, following reduction of caloric supply, we observed significant reductions in body weight, glucose intolerance, and soft tissue weights, independent of genotype (Supplemental Figure 5, A–D). Additionally, s.c. and epididymal white adipocytes were much smaller in both CR-treated control and BMAd-DTA mice (Supplemental Figure 5, E and F). Consistent with previous reports (21, 32), BMAT volume was dramatically expanded in both proximal tibial rBMAT and distal tibial cBMAT of control mice following CR. Strikingly, these effects were largely blocked by DTA expression in BMAds (Figure 4, A–D). Figure 4BMAd-DTA expression largely blocked caloric restriction–induced BMAT expansion. Control and BMAd-DTA male mice at 24 weeks of age underwent 30% caloric restriction (CR) for 12 weeks (n = 6–10 per group). Tibiae were collected for BMAT quantification. – indicates ad libitum, + indicates CR. (A and B) Decalcified tibiae were stained with osmium tetroxide. 3D images (A) and quantitative data of BMAT (B) were collected from μCT analyses. (C and D) Calcified (C) or decalcified tibiae (D) were plastic- or paraffin-processed and sectioned for Goldner’s trichrome (proximal tibiae; C) or H&E staining (distal tibiae; D). Images were taken under 100× magnification. Scale bar: 200 μm. Goldner’s trichrome staining, red-hematopoietic cells; green-bone, circular void-BMAds. Data are presented as mean ± SD. *P < 0.05 with 2-way ANOVA analyses followed by Šídák’s multiple-comparison test. It is important to note that, since CR treatment was initiated in adult mice, long bone length was not affected in either genotype (Supplemental Figure 5G). Although rBMAT was dramatically enhanced by CR in control mice, CR-induced trabecular bone loss has mainly been noted at younger ages (3, 33); thus, we were not surprised to find little difference in trabecular bone variables in our cohorts (Supplemental Figure 5H). Interestingly, 12 weeks of CR in adult mice was sufficient to cause distal tibial cortical bone loss in controls, which was accompanied by a significant expansion of cBMAT toward the tibia-fibula junction. DTA expression in BMAds depleted cBMAT under standard and CR treatments and protected BMAd-DTA mice from cortical bone reduction with CR (Figure 5, A and B), without significant effects on total bone volume in distal tibiae (Supplemental Figure 5I). Dynamic histomorphometry analysis revealed that, following CR, BMAd-DTA mice had significantly increased Ir. L. Wi, double-labeled perimeter (dL. Pm), and mineralizing proportion (M. Pm/Ec. Pm), and a tending increase of mineralizing perimeter (M. Pm) at the endosteal (Ec.) bone surface (Figure 5, C and D), without affecting total endocortical perimeter (Ec. Pm) and single-labelled perimeter (sL. Pm) (Supplemental Figure 5J). In contrast, periosteal bone surfaces were minimally labeled in most CR mice, regardless of genotype (Figure 5C). In addition to enhanced endocortical bone formation, circulating bone resorption marker CTX-1 was significantly less in CR BMAd-DTA mice compared with ad libitum BMAd-DTA mice (Supplemental Figure 5K), which may partially contribute to the resistance of CR-induced cortical bone loss. CR did not affect trabecular bone parameters in caudal vertebrae from control mice; moreover, the increase of caudal vertebral bone in BMAd-DTA mice persisted, regardless of diet (Figure 5, E and F). Figure 5Loss of BMAds protects caloric restricted mice from loss of cortical bone. Control and BMAd-DTA male mice at 24 weeks of age underwent 30% caloric restriction (CR) for 12 weeks (n = 6–10 per group). Tibiae and caudal vertebrae were collected. – indicates ad libitum, + indicates CR. (A and B) Distal tibial cortical bone area fraction (Ct. BA/TA) and mineral density (Ct. BMD) were determined by μCT scanning. Scale bar: 500 μm. (C and D) Calcified distal tibiae were cross-sectioned for dynamic histomorphometry. Endosteal bone formation was measured. Ec, endocortical; Ir. L. Wi, interlabel width; dL. Pm, double-labeled perimeter; M. Pm, mineralizing perimeter. (E and F) Caudal vertebral trabecular bone variables were analyzed following μCT scanning. Scale bar: 200 μm. Tb., Trabecular bone; BV/TV, bone volume fraction; BMD, bone mineral density; Conn. Dens, connective density; N, number; Th, thickness; Sp, separation. Data are presented as mean ± SD. *P < 0.05 with 2-way ANOVA analyses followed by Šídák’s multiple-comparison test. cBMAT depletion protects female mice from OVX-induced cortical bone loss. To further evaluate effects of BMAT loss on bone homeostasis, female BMAd-DTA mice underwent sham or OVX surgery. Surprisingly, uterine weights were higher in BMAd-DTA mice, but OVX uniformly decreased uterine weights, as expected (Supplemental Figure 6A). We observed an effect of OVX on body weight by 3-way ANOVA, due to a small and transient increase in body weight at postoperative weeks 2 and 3 (Supplemental Figure 6B). OVX BMAd-DTA mice ultimately developed slightly impaired glucose tolerance, despite similar soft tissue weights, including sWAT, eWAT, liver, and spleen (Supplemental Figure 6, C and D). Consistent with previous studies (34, 35), OVX increased both rBMAT and cBMAT, whereas BMAd-specific DTA expression depleted BMAT at baseline and blocked OVX-induced expansion (Figure 6A). OVX stimulated a slight loss of proximal tibial trabecular bone in control and caused a significant reduction in BMAd-DTA mice (Supplemental Figure 6E). In contrast, whereas OVX induced loss of cortical bone area (Ct. BA/TA) and Ct. BMD in the middiaphysis of control mice, depletion of cBMAd increased cortical bone at baseline and protected mice from cortical bone loss with OVX (Figure 6, B and C). OVX also increased total cortical bone volume in control mice but not in BMAd-DTA mice (Supplemental Figure 6F). Although there was a trend for the circulating osteoblast marker osteocalcin to be increased by OVX and/or BMAT depletion, there were no changes in the bone resorption marker CTX-1 (Supplemental Figure 6G). Dynamic histomorphometry analyses of cortical bone revealed an increase in Ir. L. Wi in the endosteum of BMAd-DTA sham mice (Supplemental Figure 6, H and I); however, further studies will be required to explain the protective effects of BMAd-DTA mice in response to OVX. Trabecular bone of caudal vertebrae was not significantly altered by OVX in either genotype; however, BMAd-DTA mice had higher bone volume fraction (Tb. BV/TV), BMD (Tb. BMD), trabecular number (Tb. N), and tighter separation between trabeculae (Tb. Sp) when compared with controls (Figure 6, D and E). These data suggest that rBMAT and cBMAT may have opposing effects on bone homeostasis under conditions of estrogen deficiency. Figure 6BMAd depletion protects mice from ovariectomy-induced cortical bone loss. Control and BMAd-DTA female mice at 20 weeks of age underwent ovariectomy (OVX). Mice were euthanized 6 weeks after surgery (n = 4–7 per group). Tibiae and caudal vertebrae were collected. – indicates sham, + indicates OVX. (A) Decalcified tibiae were paraffin embedded, sectioned, and H&E stained. Representative images from proximal and distal tibiae were collected under 100× magnification. Scale bar: 200 μm. (B and C) Tibiae were used for μCT scanning. Cortical bone area (Ct. BA/TA) and mineral density (Ct. BMD) at midtibia shaft were quantified. Scale bar: 200 μm. (D and E) Caudal vertebrae were scanned by CT. Trabecular bone volume (Tb. BV/TV) and mineral density (Tb. BMD), as well as microstructure parameters, were measured. Scale bar: 200 μm. Tb. N, trabecular number; Tb. Sp, trabecular separation. Data are presented as mean ± SD. *P < 0.05 with 2-way ANOVA analyses followed by Šídák’s multiple-comparison test. BMAT deficiency promotes bone healing. To investigate potential effects of BMAd depletion on bone repair, we induced distal tibial fractures as previously published (36) (Figure 7A). We collected tibiae at postfracture days 10 and 20 and used μCT scanning to visualize and evaluate callus formation (Figure 7, B and C). Interestingly, at day 10, the bone formation marker total procollagen type 1 N-terminal propeptide (P1NP) was increased in circulation of BMAd-DTA relative to control mice; however, P1NP concentrations were comparable between genotypes by day 20. Of note, the bone resorption marker CTX-1 remained unchanged between genotypes (Figure 7D), suggesting that BMAT depletion promotes bone formation during early stages of the fracture repair process. It was challenging to evaluate callus mineralization and bone volume fraction at postfracture day 10, given that calluses were largely composed of fibrocartilage at this time point (Figure 7, E–G). However, by postfracture day 20, fractures have mostly gone through endochondral ossification, and there is very little cartilage present. We observed a significantly smaller callus size in female BMAd-DTA mice (Figure 7, H and I). Of note, these calluses tended to have higher BMD and more dense mineralization within the bony portion (tissue mineral density [TMD]) (Figure 7J), suggesting that female BMAd-DTA mice more rapidly form higher-quality mineralized bone than control mice. In male mice, overall callus size was similar between genotypes, but BMAd-DTA mice developed higher-quality calluses with increased bone volume fraction (BV/TV) and BMD (Figure 7, K–M). Figure 7BMAd depletion promotes bone repair. (A–H) Control (–) and BMAd-DTA (+) female mice at 24 weeks of age underwent surgery to fracture distal tibiae. Tibiae and serum were collected 10 or 20 days after surgery (n = 12–15 per group; mice were split into day 10 or 20 euthanization after surgery). (A) Distal tibial fracture was visualized by x-ray scanning during surgery. (B) Representative 3D reconstruction images of tibial callus after 10 or 20 days of healing. (C) Longitudinal and cross-sectional images of callus formation from μCT analyses are shown. (D) Circulating bone formation marker (P1NP) and resorption marker (CTX-1) were analyzed by ELISA. Data are expressed as mean ± SD. *P < 0.05 with 2-way ANOVA analyses followed by Šídák’s multiple-comparison test. (E–G) Safranin O fast green (SOFG) staining and μCT analysis using Dragonfly software were performed to analyze callus formation in tibiae collected from day 10 of fracture healing. Green, bone tissue; orange, cartilage; dark blue, nuclei. Callus sizes and cartilage area percentage were quantified by ImageJ (NIH) (F). (H–J) Tibiae collected at day 20 following fracture were used for callus analysis. (K–M) Control (–) and BMAd-DTA (+) male mice at 24 weeks old underwent distal tibial fracture and were euthanized at day 20 after procedure (n = 8–9 per group). Callus formation was analyzed by SOFG staining and μCT scanning. TV, total volume; BV, bone volume; BV/TV, bone volume fraction; BMD, bone mineral density; TMD, tissue mineral density. Data are presented as mean ± SD. *P < 0.05 with a 2-tailed t test. Multiple unpaired t tests were performed, and P values were adjusted for multiple comparisons using 2-stage step-up (Benjamini, Krieger, and Yekutieli) with FDR method. Scale bar: 1 mm. BMAd-specific Pparg deficiency recapitulates bone phenotypes observed in BMAd-DTA mice. Since DTA-induced cell death may conceivably cause tissue damage by activating inflammation or other secondary responses, we also generated a mouse model of BMAd-specific Pparg KO to impair adipogenesis or stimulate death of BMAd (37, 38). BMAd-Pparg–/– female mice did not exhibit differences in body weight, random glucose concentration, or soft tissue weights (Supplemental Figure 7, A and B), whereas rBMAT and cBMAT were significantly decreased (Figure 8, A–C), confirming specificity and effectiveness of Pparg KO. BMAT loss did not affect bone growth, as evidenced by similar body and tibial lengths between BMAd-Pparg–/– mice and control littermates (BMAd-Pparg+/+) (Supplemental Figure 7C). However, cBMAT-enriched distal tibiae had higher cortical bone area (BA/TA) and thickness (Th.) (Figure 8, D and E) without significant changes in total bone or marrow volume (Supplemental Figure 7D). Although the circulating bone formation marker P1NP was increased, and proximal tibial rBMAT was largely depleted, trabecular bone parameters were not influenced (Supplemental Figure 7, E and F). Similar to observations in BMAd-DTA mice (Figure 3, A and B), cBMAT was mildly decreased in caudal vertebrae of BMAd-Pparg–/– mice (Figure 8F), and trabecular bone volume fraction (BV/TV) and BMD of caudal vertebrae were significantly higher (Figure 8, G and H). Similar results were observed in male BMAd-Pparg–/– mice. No differences were observed for body weight, random glucose, and soft tissue weights, but an increase in trabecular bone parameters of caudal vertebrae was observed in male BMAd-Pparg–/– mice (Supplemental Figure 8, A–F). We next used long bones of BMAd-Pparg male mice to further investigate mechanisms. Gene expression analysis of distal tibiae revealed upregulation of bone formation markers, such as Sp7 and Bglap (Figure 8I). Dynamic histomorphometry of distal tibiae of BMAd-Pparg–/– mice demonstrated increased double-labeled perimeter (Ec. dL. Pm), total mineral perimeter (Ec. M. Pm) and mineralizing surface per endosteal surface (Ec. M. Pm / Ec. Pm), whereas endosteal total perimeter (Ec. Pm), Ir. L. Wi, and single-labelled perimeter (Ec. sL. Pm) were not significantly changed (Figure 8, J and K, and Supplemental Figure 8G). Furthermore, these parameters were not altered on the periosteal surface (Figure 8L and Supplemental Figure 8H), providing further support for the conclusion that cBMAT depletion specifically increases endosteal bone formation. Figure 8BMAd-Pparg deficiency recapitulates bone phenotypes observed in BMAd-DTA mice. Female mice with excision of exons 1 and 2 of Pparg in BMAds (BMAd-Pparg–/–) and their littermate controls (BMAd-Pparg+/+) were housed until 20 weeks of age (n = 5 per group). Tibiae and caudal vertebrae were collected. +/+ indicates control, –/– indicates BMAd-Pparg KO. (A and B) Tibiae were decalcified and stained with osmium tetroxide for 48 hours. μCT scanning was performed. Representative 3D reconstitution images of tibiae are shown (A). BMAT in proximal (rBMAT) and distal tibiae (cBMAT) were quantified and normalized by total volume (TV) (B). (C) Decalcified bones were processed for paraffin sectioning and H&E staining. Images were taken under 100× magnification. Scale bar: 200 μm. (D and E) Distal tibial cortical bone area (Ct. BA/TA) and thickness (Ct. Th) were quantified following μCT scanning. Scale bar: 500 μm. (F) Decalcified caudal vertebrae were paraffin embedded, sectioned, and H&E stained. Representative pictures were taken under 100× magnification. Scale bar: 200 μm. (G and H) Trabecular bone (Tb.) parameters in caudal vertebrae were measured by CT. Scale bar: 200 μm. BV/TV, bone volume fraction; BMD, bone mineral density. (I–L) Distal tibiae from male mice at 24 weeks of age were collected for mechanistic analyses (n = 5 per group). (I) Distal tibial total RNA was purified and used for qPCR to measure osteogenic markers. The expression of Sp7 and Bglap genes were normalized to the geomean of Hprt and Rpl32a. (J) Distal tibiae were cross-sectioned and scanned for calcein-labeled mineralizing bone surfaces. (K and L) Dynamic histomorphometry was performed in endosteal (K) and periosteal (L) surfaces. Endocortical double-labeled perimeter (Ec. dL. Pm) and mineralizing perimeter (Ec. M. Pm) were quantified by BioQuant software. These parameters were also quantified at periosteal (Ps.) mineralizing surfaces. Data are presented as mean ± SD. *P < 0.05 with a 2-tailed t test.
|
|
Scooped by
Gilbert C FAURE
October 18, 2022 7:00 AM
|
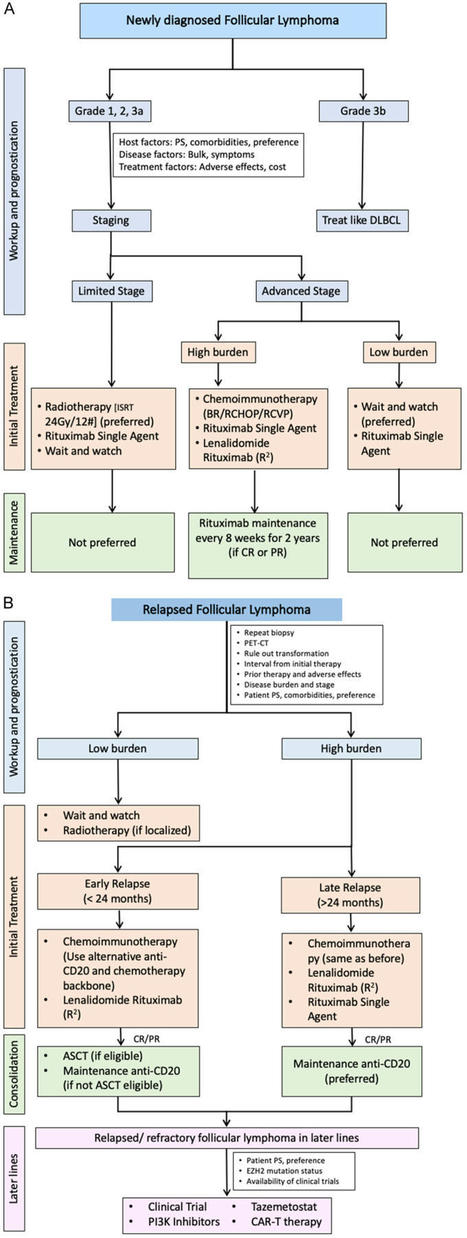
High-dose chemotherapy (HDCT) followed by an autologous stem cell transplant (ASCT) is the standard of care for most patients who relapse following initial therapy.For patients who fail HDCT with ASCT, brentuximab vedotin, PD-1 blockade, non-myeloablative allogeneic transplant, or participation in...
|
|
Scooped by
Gilbert C FAURE
September 28, 2022 12:30 PM
|
Bonjour à toutes et tous,
fier et ravi de vous annoncer qu'aujourd'hui parait l'ouvrage "Hématologie" de la collection "Objectif Internat Pharmacie" coordonné…
|
|
Scooped by
Gilbert C FAURE
September 23, 2022 10:28 AM
|
Abstract IgM monoclonal gammopathy of undetermined significance is a pre-malignant condition for Waldenström macroglobulinemia and other B-cell malignancies, defined by asymptomatic circulating IgM monoclonal protein below 30 g/L with a lymphoplasmacytic bone marrow infiltration of less than 10%. A significant proportion, however, develop unique immunological and biochemical manifestations related to the monoclonal protein itself in the absence of overt malignancy and are termed IgM-related disorders or, more recently, monoclonal gammopathy of clinical significance. The indication for treatment in affected patients is dictated by the pathological characteristics of the circulating IgM rather than the tumor itself. The clinical workup and treatment options vary widely and differ from those for Waldenström macroglobulinemia. The aim of this review is to alert clinicians to IgM monoclonal gammopathy of clinical significance and to provide practical guidance on when to screen for these phenotypes. We discuss clinical characteristics, the underlying clonal profile, diagnostic workup and treatment considerations for five important subtypes: cold agglutinin disease, type I and II cryoglobulinemia, IgM-associated peripheral neuropathy, Schnitzler syndrome and IgM-associated AL amyloidosis. The inhibition of the pathogenic effects of the IgM has led to great success in cold agglutinin disease and Schnitzler syndrome, whereas the other treatments are centered on eradicating the underlying clone. Treatment approaches in cryoglobulinemia and IgM-associated peripheral neuropathy are the least well developed. A multidisciplinary approach is required, particularly for IgM-related neuropathies and Schnitzler syndrome. Future work exploring novel, clone-directed agents and pathogenic IgM-directed therapies is welcomed. IntroductionIgM monoclonal gammopathy of undetermined significance (MGUS) is defined by asymptomatic circulating IgM monoclonal (M) protein below 30 g/L with a lymphoplasmacytic bone marrow infiltration of less than 10%.1 IgM MGUS is a pre-malignant condition for non-Hodgkin lymphomas, mostly Waldenström macroglobulinemia (WM), chronic lymphocytic lymphoma, and plasma cell neoplasms. Most patients are candidates for observation. However, some develop diverse immunological and biochemical manifestations related to the monoclonal protein itself.2 This may lead to organ damage, even in the absence of overt malignancy. These so-called IgM-related disorders are a distinct clinical entity termed monoclonal gammopathy of clinical significance (MGCS).3 The clinical workup and treatment options vary widely and differ from those for WM, which have been outlined in recent consensus guidelines.4The aim of this review is to alert clinicians to IgM MGCS and to provide practical guidance on when to screen for these phenotypes. We discuss the clinical characteristics, diagnostic workup and treatment considerations for five important subtypes: cold agglutinin disease (CAD), cryoglobulinemia, IgM-associated AL amyloidosis, IgM-related neuropathies and Schnitzler syndrome. A comprehensive list of IgM MGCS is listed below (Table 1).Clonal characterizationIt is important to identify the underlying clone as IgM MGUS may progress to a number of lymphoproliferative disorders or very rarely to myeloma.5 IgM MGUS most commonly arises from a CD20+ lymphoplasmacytic cell without class-switching.1 The risk of progression to lymphoma, chronic lymphocytic leukemia, AL amyloidosis or multiple myeloma is 1.1 event per 100 person-years.5 In the largest series of 210 patients with IgM MGUS with a median follow-up of 29.3 months, no patients progressed to IgM myeloma.5 The incidence and prevalence of IgM MGCS are unknown. Clonal B cells in MGUS have the same genetic and molecular signature as the WM clone. However, MGUS cases have a significantly lower number of mutations than in WM, indicating multiple genetic hits are required for progression. The somatic MYD88L265P mutation constitutively activates nuclear factor kB and triggers B-cell proliferation. It is considered an early acquired mutation and is present in the majority of patients with WM or IgM MGUS.6,7 The gene encoding the chemokine receptor CXCR4, involved in homing of B cells in the bone marrow, is mutated (CXCR4MUT) in a smaller proportion. This is usually a subclonal mutation and likely a late event. IgM myeloma has a distinct cell of origin, a pro-B cell, with frequent t(11;14), an absence of MYD88L265P mutation and high BCL2/BCL2L1 ratio.8 These clonal characteristics may have therapeutic implications. Table 2 summarizes the data on the underlying histology and clonal characteristics of IgM MGCS compared with those seen in WM and IgM MGUS in general.Primary cold agglutinin diseaseIn primary CAD, autoimmune hemolytic anemia is caused by a cold agglutinin that is a monoclonal IgMk in more than 90% of cases and is produced by clonal lymphocytes in the bone marrow. The antibody binds erythrocyte antigens (typically type I) optimally at 4˚C resulting in agglutination and classical complement pathway activation.9 The thermal amplitude describes the temperature range at which the antibodies are active, and only those with a thermal amplitude reaching higher than 28˚C are considered pathogenic. In most cases, complement activation is incomplete and extravascular hemolysis of C3b-opsonized erythrocytes occurs in the liver. Less frequently there is initiation of the terminal pathway, assembly of the membrane attack complex (C5b-C9) and intravascular hemolysis, which can lead to acute life-threatening anemia. Cold agglutinins in the context of infection, autoimmune disease and overt lymphoma9 (including chronic lymphocytic lymphoma, diffuse large B-cell lymphoma and WM) are referred to as cold agglutinin syndrome. The management of cold agglutinin syndrome is directed at treating the underlying cause and is not further discussed here.Clinical characteristicsPatients with CAD present with chronic anemia and/or cold-induced circulatory symptoms. Of 232 patients in an international retrospective case series, the median IgM was 3.2 g/L and over 90% had hemolytic anemia and circulatory symptoms. Thirty-eight percent required transfusions at or before diagnosis and 47% during follow-up. Around half had acrocyanosis or Raynaud syndrome affecting daily living. Ulcers or gangrene were rare (<2%).10 In a third of cases, hemoglobin concentration is below 80g/L.11 Circulatory symptoms do not correlate with either the degree of anemia or the bone marrow histology.10,11 There is an increased risk of thrombosis in CAD, likely related to intravascular hemolysis,12,13 which is not correlated with the severity of the anemia.10 CAD is a chronic disease and affected patients have an estimated 16-year survival.10 Clonality has been demonstrated in approximately 80% of cases10,14 and the remainder likely require more sensitive methods to detect the pathogenic clone. The CAD clone has a distinct phenotype that differs from that of WM. MYD88L265P is rarely seen. Recurrent somatic mutations in CXCR4 (20%),10,14,15 KMT2D (69%) and CARD11 (31%) have been described.16 Recurrent chromosomal abnormalities have been identified.17 Based on bone marrow biopsies of 54 cases of CAD, the entity "CAD-associated lymphoproliferative disorder" has been defined, with typical morphology including absent plasmacytoid cells, universally restricted IGHV4-34 gene usage and lack of MYD88L265P.14 Most patients meet the criteria for MGUS and extramedullary disease is rare.10Diagnostic workupLaboratory findings are consistent with hemolysis (and may, therefore, include reticulocytosis, elevated lactate dehydrogenase, unconjugated hyperbilirubinemia and decreased haptoglobin), the monospecific direct antiglobulin test is strongly positive for C3d and there is a cold agglutinin titer of ≥1:64 at 4˚C. Blood samples should be handled warm until separation to prevent agglutination.TreatmentTreatment goals are to alleviate cold-induced symptoms and hemolytic anemia. Response assessment should evaluate hemolytic activity and symptoms as well as clonal response. There are no standard criteria to assess response of cold-induced peripheral symptoms and instead clinicians depend on patient-reported outcomes. A treatment algorithm is provided in Figure 1 and Online Supplementary Table S1. All patients should avoid exposure to cold and be observed, particularly during periods of febrile illness and surgery.10 Red blood cells should be transfused via a blood warmer. Symptomatic patients should commence use of folic acid and be considered for thromboprophylaxis. It is important to note that steroids and splenectomy are not effective in CAD.9,12We recommend a frontline clone-directed approach (Figure 1), although achieving complete eradication is rare. The most established treatment is rituximab-based therapy. Prospective trials of rituximab monotherapy show a modest response rate of 50% with rare complete responses.18 Real-world data show a 15-month median response duration and repeated responses in over a third of patients.10 Efficacy is greatly improved by the addition of bendamustine. In a prospective study of the bendamustine-rituximab combination with a reduced dose of 70 mg/m2 bendamustine delivered for four cycles to 45 patients, the response rate was 71%, with 40% complete responses and a median increase of hemoglobin of 44 g/L. Grade 4 neutropenia was observed in 20% of the patients and 29% required a dose reduction.19 According to updated data, both the overall and complete response rates improved due to deeper responses over time.10 Rituximab-fludarabine is efficacious (response rate: 62%; complete responses: 38%) but associated with an increased risk of secondary malignancy and is therefore not a preferred option.10,20 Based on a prospective study of 19 patients,21 the response rate to bortezomib-based treatment was 32%, although this was after only a single course of bortezomib. Bruton tyrosine kinase (BTK) inhibitors were effective in all four treated patients with relapsed CAD in a retrospective report.22 There is a case report of the use of daratumumab in CAD.23Clinical trials should be considered in relapsed disease. Promising studies have examined proximal complement inhibition to inhibit extravascular hemolysis. Complement inhibition necessitates indefinite treatment and fails to reduce vascular symptoms. In a phase III study of anti-C1s, sutimlimab, versus best supportive care, it was seen that the complement inhibitor rapidly halted hemolysis, produced transfusion independence in 73% of patients, increased hemoglobin concentration by more than 15 g/L and improved fatigue;24 this drug has now been approved by the Food and Drug Administration.24 The effect of complement inhibition on thrombosis has not been established; however, D-dimer and thrombin-antithrombin complex levels decreased on treatment.13,24 Use of the C5 inhibitor eculizumab rapidly abrogates the terminal complement pathway with a short time to response. However, in a phase II trial there was a marginal hemoglobin rise of 8 g/L.13 Proximal complement inhibition presumably has greater effectiveness because it targets C3-mediated hemolysis via the liver, which is often predominant in CAD. Ongoing clinical trials of complement inhibition in CAD include those studying the C3b inhibitor, pegcetacoplan (phase III, NCT05096403), the complement factor B inhibitor, iptacopan (phase II, NCT05086744), the C1 esterase inhibitor, cinryze (phase II, 2012-003710-13/NL) and the C1s inhibitor BIVV020 (phase Ib, NCT04269551).Acute life-threatening intravascular hemolysis may necessitate transfusion. Plasma exchange may be employed provided that all priming fluids and the circuit apparatus are pre-warmed and that the replacement products are run through a warmer. Erythropoietin support can be considered as erythropoietin can be inappropriately low in autoimmune hemolytic anemia.25 Complement-directed therapy may act as a bridge for rituximab combinations to target the underlying clone, which can take weeks to have an effect.CryoglobulinemiaCryoglobulinemia is characterized by immunoglobulins that precipitate at temperatures below 37°C and redissolve on warming. Monoclonal IgM can be associated with type I and type II cryoglobulinemia. Type I cryoglobulinemia consists of monoclonal immunoglobulins only. In type II “mixed” cryoglobulinemia there is a monoclonal component possessing avidity for the polyclonal component of a different isotype (most frequently IgM with rheumatoid factor activity, the ability to bind to the Fc portion of IgG). The rheumatoid factor detected in type II cryoglobulinemia is a monoclonal IgMk in over 85% of cases.26 While most cases of type II cryoglobulinemia are related to hepatitis C, here we focus on those related to monoclonal IgM.Clinical characteristicsData characterizing patients with monoclonal IgM and cryoglobulinemia are scant. The clinical characteristics have been gleaned from retrospective cohorts grouping together IgG and IgM cases. The largest series reported over 1,600 unselected patients with cryoglobulinemia. Nine percent had type I cryoglobulinemia and 47% had type II.26 The only series characterizing the symptoms of IgM type I cryoglobulinemia included 26 patients; 35% had underlying MGUS, 35% had WM and 31% had non-Hodgkin lymphoma.27 The incidence is likely underestimated as most literature reports are derived from centers that do not routinely screen for cryoglobulinemia upon recognition of an IgM clone.3A wide spectrum of symptoms may be present (Figure 2). The symptoms of type I cryoglobulinemia are caused by vascular occlusion whereas those of type II are due to small and medium vessel vasculitis. Cutaneous involvement is most frequent in type I cryoglobulinemia. Cutaneous manifestations range from purpura, livedo reticularis, acrocyanosis to cold urticaria, digital ischemia, ulcers and necrosis. Among 26 patients, 46% had skin involvement and less than 10% had peripheral neuropathy (8%), arthralgia (8%) or renal involvement (4%).27 Other studies found peripheral neuropathy in a higher proportion of IgM cases, mainly sensory neuropathy (70%), but sensorimotor polyneuropathy and mononeuritis multiplex were also seen.28 Central nervous system involvement is rare unless due to hyperviscosity.29 No studies have reported specific presenting features of type II cryoglobulinemia in patients with circulating monoclonal IgM. In a mixed cohort of 203 type II patients with an underlying hematologic disorder in 23%, skin manifestations predominated (85%). Compared to type I cryoglobulinemia there was a greater proportion of peripheral neuropathy (56%), joint (41%), renal (38%), gastrointestinal (6%) and pulmonary (2%) involvement. Hyperviscosity is almost never seen.Diagnostic workupLaboratory testing is critical as a minimal amount of measurable cryoglobulin may cause symptoms. In one study in which two-thirds of patients were symptomatic, 58% of the IgM type I cryoglobulinemia cases had a cryocrit of <1%, which was a significantly greater proportion than in IgG cryoglobulinemia.27 Symptoms do not correlate with the cryocrit and depend instead on the temperature at which precipitation occurs.29 Accurate detection of cryoglobulins requires samples to be taken into prewarmed tubes which must not be allowed to cool below 37˚C until the serum is separated, as the cryoglobulin may precipitate and not be detected. Similarly, a false-negative M-protein result may result from the same process. In a French study, 9% of cases with negative results were positive on a follow-up test.26 Care must be taken with preanalytical variables; repeat testing of M-protein and cryoglobulins is indicated if the clinical suspicion is high. Increased plasma viscosity in the absence of a high IgM should trigger clinicians to consider cryoglobulinemia.A tissue biopsy may be indicated to identify renal or nerve involvement and distinguish it from other causes. Intravascular precipitation of IgM triggered by exposure to cold results in thrombotic obstruction and ischemia in small vessels as evidenced on biopsy in type I cryoglobulinemia. Leukocytoclastic vasculitis may be evident in type II cryoglobulinemia.TreatmentA treatment approach is outlined in Figure 2. There is a paucity of data to guide optimal management. Mild symptoms may abate with cold prevention. Rapidly progressive nephropathy and neuropathy have been reported at various stages of the disease course, so careful monitoring is recommended.28 When cryoglobulinemia is tested for exclusively in symptomatic patients, treatment is commenced for cryoglobulinemia-related symptoms in the majority (80%).30 Response assessment is not standardized and mostly focuses on symptomatic improvement.27 The cryocrit at treatment initiation, change in cryocrit and time to nadir were predictive of symptom improvement in a mixed cohort of patients with IgG and IgM type I cryoglobulinemia. The underlying diagnosis of MGUS or lymphoma did not affect symptom improvement.30Treatment regimens are heterogeneous and have been used in small series of patients. Plasma exchange may temporize critical symptoms and is used in up to a third of all cases of cryoglobulinemia in mixed cohorts; a warming procedures should be in place.30-32 In the absence of robust evidence, definitive treatment should be directed at the underlying clone. Steroids (1 mg/kg) are used in up to 90% of all cases of cryoglobulinemia, often together with immunosuppression.31,32 Rituximab combinations or bortezomib-based treatment are typically employed27 with symptomatic responses in approximately 80% of cases.27,29,31 Disappearance of cryoglobulin may be seen in half of patients.30 Transient disease exacerbation (an ‘IgM flare’) has been described following the use of rituximab in type I cryoglobulinemia with a low disease burden (<10% infiltrate)33 and in type II cryoglobulinemia.34 Some authors have suggested that a post-rituximab flare in type II cryoglobulinemia may be due to the exogenous IgG from the rituximab infusion which may also be a target of the monoclonal IgM. A study examining plasma exchange prior to rituximab to prevent IgM flares is ongoing (NCT04692363). Currently there are no data on the use of autologous stem cell transplantation or BTK inhibitors in IgM-associated cryoglobulinemia.IgM-associated AL amyloidosisAL amyloidosis is a rare disorder caused by extracellular deposition of insoluble misfolded monoclonal light chain fragments, produced by an underlying plasma cell dyscrasia or lymphoma, as amyloid fibrils in tissues. IgM-associated amyloidosis accounts for 5 to 7% of all systemic amyloidoses.35-39 In non-IgM AL amyloidosis, advances in treatment have resulted in marked improvement in survival, although patients with advanced disease have a poor outcome. Data on IgM-associated AL amyloidosis show no improvement over time.40Clinical characteristicsDue to its rarity, IgM-associated amyloidosis is less well characterized but increasingly recognized as a distinctive entity.40 When compared to non-IgM amyloidosis, patients are older36,37 with a history of MGUS or WM up to 65 months prior to diagnosis.38Multiple series36,38,41 indicate a smaller proportion of λ light chain involvement compared to that in non-IgM cases. Presenting free light chain levels are lower than in non-IgM AL amyloidosis and in the largest study so far of IgM-associated amyloidosis only two-thirds of the 250 patients had a greater than 50 mg/L difference between involved and uninvolved free light chains.40 The pattern of organ involvement is also different, with a greater propensity for lymph node and soft tissue deposition (35%). Cardiac involvement is less common (45%) and neuropathy more frequent (28%).36,38,40Diagnostic workupThe exact nature of the clonal dyscrasia in IgM-associated AL amyloidosis remains unclear. The Mayo group has suggested two types, based on morphology; lymphoid predominant (lymphoplasmacytic lymphoma) or plasma cell predominant (pure plasma cell neoplasm).38 Of 75 cases, the lymphoid predominant type (63%) showed a higher tumor infiltrate, MYD88L265P in 84%, CXCR4MUT in 29% but absent t(11;14), similar to WM. By contrast, the cases of pure plasma cell neoplasm (23%) had similar rates of t(11;14) compared to non-IgM-associated amyloidosis and no MYD88L265P/CXCR4MUT, similar to IgM myeloma.38 Patients with the pure plasma cell neoplasm type appear to have poorer outcomes. These findings need independent confirmation to hone treatment approaches.TreatmentThere are no consensus guidelines, approved treatments or prospective clinical trials for IgM amyloidosis. The aims of treatment are to reduce the clonal burden and improve performance status with a view to extending survival. A treatment algorithm is summarized in Figure 3. Evidence is largely limited to retrospective series with heterogeneous regimens. Criteria developed for response assessment in non-IgM AL amyloidosis are applicable to IgM-associated AL amyloidosis with assessment of hematologic response and organ response. Response assessment by both free light chains and M-protein had prognostic significance in retrospective series35,36 alongside age, Mayo stage, cardiac involvement, liver involvement40 and prior WM treatment.38 β2-microglobulin and lactate dehydrogenase levels do not independently affect survival,41 unlike in WM,42 which may be related to the low tumor burden. Despite less cardiac involvement, patients with IgM-associated amyloidosis do not have superior survival compared to those with non-IgM-associated amyloidosis,38 attributable to the inability to achieve deep clonal responses.Induction of hematologic response is more challenging with a reported 6-month overall response rate of 39% versus 59% (P=0.008), deep responses are seen in only 24%.38Organ response rates are consequently poor (5% cardiac, 18% renal) and lower than those in a non-IgM-associated cohort.43Strategies to target the lymphoplasmacytic and plasma cell clones have been employed. The best outcomes have been achieved by autologous stem cell transplantation, with more than 90% achieving a hematologic response.40,41,44 However, up to just 25% of all-comers were eligible for this intense therapy. The largest series of autologous stem cell transplantation in 38 patients44 included 58% who had received prior therapy and the 100-day mortality was 5%. There was, however, a relatively low rate of cardiac involvement (26%), demonstrating the importance of the selection of patients. Induction chemotherapy prior to autologous stem cell transplantation is not universally utilized. Conditioning most commonly involves melphalan, however the BEAM (carmustine, etoposide, cytarabine, melphalan) regimen has also been used.44As the majority of cases have an underlying lymphoplasmacytic clone, induction therapy with rituximab-based combination chemotherapy is strongly preferred. In 27 cases, the bendamustine-rituximab combination resulted in an intention-to-treat hematologic response rate of 59%, with complete responses in 11%, and a median progression-free survival of 34 months. Sixty percent of patients treated with this combination in second line achieved a very good partial response.45 Bendamustine is neither neurotoxic nor cardiotoxic. Bortezomib in combination with rituximab and dexamethasone may provide rapid disease control. The only prospective trial of this strategy recruited ten patients over 1 year.46 A hematologic response was achieved by 78% with sustained responses at a median of 11 months, after only two cycles. However, there were no complete responses. Treatment had to be interrupted in 30% of patients because of toxicity. Patients with grade 3 sensory and/or grade 1 painful neuropathy were excluded and treatment-related neuropathy is a particular concern in these patients. Responses to frontline alkylating agents have been disappointing. In a series of 46 patients treated after 2003, the hematologic response rate was 37% and there were no complete responses.41 Immunomodulatory drugs alone result in variable response rates, but mostly less than 50%. BTK inhibitors, although promising in WM, have been associated with low response rates in IgM-associated amyloidosis. Of eight patients treated with ibrutinib, only two achieved a hematologic response and the median overall survival was 9 months.47 No studies have examined anti-CD38-bortezomib combinations, which is the standard of care in non-IgM AL amyloidosis.48We consider upfront bendamustine-rituximab the treatment of choice in IgM-associated amyloidosis, consolidated with autologous stem cell transplantation when the patient’s performance status allows. There is no consensus regarding less fit patients; treatment choices need to be individualized depending on affected organs and tolerance of treatments. Overall in this condition, deep responses remain poor. Future studies are required to address whether regimens based on novel agents (including venetoclax, daratumumab and the newer BTK inhibitors) may lead to improvements in the outcomes of patients with non-IgM AL amyloidosis.IgM-related neuropathiesIgM-related peripheral neuropathies encompass an array of entities including immune-mediated neuronal damage, such as that caused by antibodies to myelin-associated glycoprotein (MAG), or direct neurotoxicity with infiltration by lymphoma (neurolymphomatosis), light chains (amyloidosis) or cryoglobulins. Peripheral neuropathy has been found to occur in 15-30% of MGUS and WM cases,49,50 but the prevalence is likely affected by selection bias and variable neurological evaluation in patients as part of a work up of IgM M-protein. The UK registry documented 153 patients with IgM-related neuropathy, comprising antiMAG neuropathy (55%), non-MAG IgM neuropathy (35%) and less frequently (<4% each) AL amyloidosis, cryoglobulinemia, anti-ganglioside neuropathy and CANOMAD syndrome (chronic ataxic neuropathy, ophthalmoplegia, IgM M-protein, cold agglutinins and disialosyl ganglioside antibodies).50Clinical characteristicsAnti-MAG neuropathy is the most common and best-defined IgM-related neuropathy. Patients typically present with chronic-onset, distal, symmetric neuropathy, sensory ataxia and tremor. Patients may be misdiagnosed as having chronic inflammatory demyelinating polyneuropathy. It is important to correctly classify the neuropathy (Table 3) as this has significant management implications. Atypical “red flag” symptoms not consistent with anti-MAG peripheral neuropathies include acute onset, rapid tempo of symptoms, pain, dysautonomia, weight loss, and cutaneous or central nervous system signs. These should alert the clinician to consider alternate diagnoses (Figure 4). CANOMAD syndrome is a very rare chronic progressive condition associated with antiganglioside antibodies. This syndrome should be considered if there is sensory loss with ophthalmoplegia or ataxia. Bing-Neel syndrome is the term for central nervous system infiltration by lymphoplasmacytic lymphoma; consensus guidelines on its diagnosis, treatment and response criteria have been published.51 Cryoglobulinemia and amyloidosis are discussed in their respective sections.Diagnostic workupThe majority of patients with IgM-related neuropathy (>90%) have symptoms of the underlying neurological disorder at diagnosis.50 This supports the strong need for careful early evaluation of patients jointly with an expert neurologist. The presence of a peripheral neuropathy alongside a serum monoclonal IgM or anti-MAG antibody does not equal a causal relationship, since gammopathies as well as peripheral neuropathies are both increasingly prevalent with age. Patients should be tested for anti-MAG antibodies, but only high-titer antibodies are clinically relevant in the presence of a characteristic clinical picture in anti-MAG neuropathy.52 A reduction in anti-MAG titers and levels of IgM M-protein with therapy appeared to correlate with improvement in neuropathy in a retrospective analysis of 50 studies.53 Responders also had a younger age of onset.53Nerve conduction tests and electromyography are warranted and characteristically show demyelination with reduced conduction velocity, disproportionately prolonged distal motor latency and absent sural potentials. Partial motor conduction block is rare. Progressive demyelination may result in secondary axonal loss which affects the likelihood of neural recovery.52 Magnetic resonance imaging of the neuraxis and evaluation of large volumes of cerebrospinal fluid may be required if central nervous system involvement is suspected. A nerve biopsy may be needed if the diagnosis remains elusive despite systematic investigation. Comprehensive consensus guidelines provide further details.52TreatmentIn general, in anti-MAG and non-anti-MAG neuropathy, treatment should be initiated only in those with significant or progressive disability.52 The aim of treatment is to halt progression and improve neurological function, although this may potentially take months to years, even after IgM responses. Although many neurological disability scales exist, they are not available outside of specialist neurology clinics and there is no standardized method of response assessment. The use of serial validated patient-reported outcome scores (e.g., the Inflammatory Rasch-Built Overall Disability Scale) is advocated, as this can be easily undertaken in non-specialist clinics.52 An observational trial is currently recruiting with an aim to develop an IgM-specific disability scale (NCT03918421). Patients should be managed in a multidisciplinary fashion with input from neurology, hematology, physiotherapy and occupational therapy.Rituximab is widely, but inconsistently used in the setting of IgM-related neuropathies. A meta-analysis of rituximab demonstrated improvement in disability scales at 8 to 12 months and long-term efficacy was demonstrated in a third of patients.54 A transient flare of symptoms following the administration of rituximab was observed in 12% in a large series of patients with anti-MAG antitbodies.55 Steroids, intravenous immunoglobulins and plasma exchange alone do not provide long-term clinical benefit in anti-MAG neuropathy56,57 and are resource-intense, respectively. In contrast, intravenous immunoglobulins and rituximab-based regimens are effective in CANOMAD syndrome (producing partial clinical responses or better in 53% and 52% of patients, respectively),58 while chronic inflammatory demyelinating polyneuropathy is responsive to intravenous immunoglobulins,52 highlighting the relevance of correct diagnostic classification.Although data are largely limited to retrospective series, targeting the underlying clone is feasible in IgM-related neuropathy; the optimum depth of response is unknown. Clinical improvement or stabilization is significantly more likely with rituximab-containing therapy (dexamethasone, rituximab, cyclophosphamide; bendamustine plus rituxi-mab; cyclophosphamide, prednisolone, rituximab, vincris-tine), non-amyloid-related neuropathy and attainment of at least partial haematologic response.49,50There is an unmet need for reliable biomarkers for diagnosis, appropriate selection of patients for treatment and criteria for monitoring response.59 There is a lack of prospective clinical trials to optimize treatment options. A phase II clinical trial, MAGNAZ, of the oral BTK inhibitor zanubrutinib in anti-MAG peripheral neuropathies is underway.60Schnitzler syndromeSchnitzler syndrome is a rare auto-inflammatory disorder characterized by an IgM monoclonal gammopathy and chronic recurrent urticarial rash. The Strasbourg criteria outline additional minor criteria of recurrent fever, abnormal bone remodeling with or without bone pain, neutrophilic dermal infiltrate, leukocytosis and elevated C-reactive protein.61 Around 300 cases have been reported to date. It is underdiagnosed and, despite its rarity, is important to identify as specific treatment can significantly improve quality of life.62Clinical characteristicsOf 281 cases in the largest case series, fever was present in 72%, anemia in 63%, arthralgia in 68%, bone pain in 55%, lymphadenopathy in 26%, and liver or spleen enlargement and neuropathy in less than 10%.63 In smaller series fatigue and weight loss were documented in up to around 50% of cases.62,64 The urticarial rash can cover any part of the body, but face, palm and sole involvement is infrequent, as is intense pruritis. Skin lesions typically resolve within hours.65 The time from onset of symptoms to diagnosis is long, at a median of 5 years and may be as long as 20 years.62The monoclonal gammopathy is almost always IgMk. Bone marrow involvement is minimal, being around 4% in one series, and a median M-protein concentration of 6 g/L has been documented.62 In the largest case series, 63% of the 281 bone marrow samples were reported as normal.63 The MYD88L265P mutation was detected in the peripheral blood of 30% of 30 patients.66 The authors suggested that the presence of this mutation may correlate with the risk of WM, although the mutation detection rate may have been underestimated as the sensitivity of detecting peripheral blood B-cell clones may be hampered when the level of disease burden is low. The frequency of the MYD88L265P mutation in bone marrow has not been studied. Chronic inflammation may lead to AA amyloidosis in 2% of cases of Schnitzler syndrome. At a median of 8 years, the rate of evolution to lymphoma is 20%, which is in line with progression in unselected cohorts of patients with IgM MGUS.61,63Schnitzler syndrome is associated with cytokine dysregulation. It bears close phenotypic resemblance to an inherited disorder, cryopyrin-associated periodic syndrome, caused by gain-of-function mutations in the NLRP3 gene. This results in upregulation of interleukin (IL)-1b production and has informed therapeutic options in Schnitzler syndrome, by targeting IL-1b.Diagnostic workupThere is no single diagnostic test and the diagnosis is made based on clinical characteristics. Differential diagnoses for the rash and fever include adult-onset Still disease, systemic lupus erythematosus, acquired C1 esterase deficiency, cryopyrinopathies and cryoglobulinemia (coldinduced urticaria). Skin biopsy reveals a neutrophilic urticarial dermatosis without features of vasculitis.TreatmentTreatment is aimed at reducing the considerable associated morbidity related to rash, fever and joint and bone pain. Symptoms respond poorly to historic first-line agents including antihistamines, nonsteroidal anti-inflammatory drugs, dapsone and colchicine.65 The use of high-dose steroids, although moderately effective, is limited by long-term toxicities.Without anti-IL treatment, morbidity is high. In a series of 21 patients, all had almost daily symptoms with a profound effect on their quality of life.64 Anti-IL-1 agents, such as anakinra, canakinumab, and rilonacept, have all been used but not directly compared. Anakinra is the agent with which experience is greatest and is the treatment of choice. It is a recombinant IL-1-receptor antagonist and has the greatest efficacy (94% efficacy in 86 cases),63 with durable responses (83% complete responses after a median of 36 months).67 Anakinra has a half-life (t1/2) of 4-6 hours and provides impressive control of all signs within hours, normalization of C-reactive protein levels and abrogation of the risk of AA amyloidosis.64 Nonetheless, patients require continuous daily injections and relapse occurs after treatment discontinuation. Canakinumab, an IL-1b monoclonal antibody, is long-acting (t1/2 21-28 days) and is, therefore, administered less frequently. Data from phase II, placebo-controlled, randomize trials have demonstrated its efficacy. For 17 patients in a long-term study, clinical efficacy was greatest when patients injected canakinumab as needed. A systematic review of 34 patients showed that 59% achieved complete responses.68 Rilonacept, an IL-1 binding and neutralizing fusion protein, achieved near complete responses in 50% of cases.69 To cilizumab, an IL-6 receptor antagonist, has been beneficial in three patients who were refractory to anakinra.70Cyclophosphamide, rituximab and ibrutinib have achieved responses when treatment was given for overt lymphoma but have been largely ineffective or untested in the absence of lymphoma.65 There is little to support the notion that anti-IL therapy affects the underlying B-cell clone.ConclusionWe have discussed a range of distinctive entities of IgM MGCS, including their specific clinical characteristics, underlying clonal profile, and diagnostic workup as well as treatment considerations. Careful evaluation of the presenting features and thorough interrogation of the underlying clone are critical. Determining the nature of either a mature B-cell derived clone or plasma cell clone will have management implications. There is an IgMk predominance in all cases except IgM-associated AL amyloidosis. The indication for treatment is dictated by the pathological characteristics of the circulating IgM rather than by the tumor itself. While deep suppression of the pathogenic IgM is typically required for response, achieving long-term clonal eradication is challenging, as demonstrated by low complete response rates. Treatment inhibiting the pathogenic effects of IgM while not directed at the underlying clone has led to great success in CAD (complement inhibitors) and Schnitzler syndrome (cytokine inhibition), whereas the other treatments are centered on eradicating the underlying clone. Treatment approaches in cryoglobulinemia and IgM-related peripheral neuropathies are the least well developed. A multidisciplinary approach is required particularly for IgM-related neuropathies and Schnitzler syndrome.Due to the rarity of IgM MGCS, data are scant and collaborative research is imperative to aid defining optimal treatment strategies. International registries may better define characteristics and assess treatment outcomes. Future work exploring clone-directed treatment options and pathogenic IgM-directed therapies is welcomed.Footnotes Received March 9, 2022 Accepted June 16, 2022 CorrespondenceJ.M. Vos j.m.i.vos@amsterdamumc.nl Disclosures JK has no conflicts of interest to disclose. SD'S has received research funding, honoraria for advisory board work and conference support from Janssen and BeiGene, and honoraria for advisory board work from Sanofi. MCM has provided speakers bureau services for Medscape and BMS, consultancy for Janssen-Cilag and Gilead Sciences Netherlands B.V., sat on advisory boards for Janssen Pharmaceutica and Alnylam, and received hospitality support from Celgene. MJK has acted as a consultant for and received honoraria and travel support from Novartis and Miltenyi Biotec, has received research funding from Takeda and Celgene, has acted as a consultant for and received honoraria, travel support and research funding from Kite, a Gilead Company, and Roche, and has acted as a consultant for and received honoraria from BMS/Celgene. AW has provided consultancy services for Alexion, AstraZeneca Rare Diseases and Janssen, and has received honoraria from Celgene and Takeda, clinical trial funding from Caelum Biosciences, and research funding from Amgen. JMIV has received reimbursement of travel costs from Celgene, receives research funding from Beigene (institutional) and has participated in an advisory board as well as acted as a consultant for Sanofi (institutional honoraria) Contributions JK and JMIV wrote the first draft of the manuscript, SD’S, MCM, MJK, and AW equally reviewed and added critical discussions throughout the writing of the review. References Swerdlow SH, Campo E, Pileri SA. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016; 127(20):2375-2390. Google Scholar Fermand JP, Bridoux F, Dispenzieri A. Monoclonal gammopathy of clinical significance: a novel concept with therapeutic implications. Blood. 2018; 132(14):1478-1485. Google Scholar Owen RG, Treon SP, Al-Katib A. Clinicopathological definition of Waldenstrom's macroglobulinemia: consensus panel recommendations from the Second International Workshop on Waldenstrom's Macroglobulinemia. Semin Oncol. 2003; 30(2):110-115. Google Scholar Castillo JJ, Advani RH, Branagan AR. Consensus treatment recommendations from the tenth International Workshop for Waldenström Macroglobulinaemia. Lancet Haematol. 2020; 7(11):e827-e837. Google Scholar Kyle RA, Larson DR, Therneau TM. Long-term follow-up of monoclonal gammopathy of undetermined significance. N Engl J Med. 2018; 378(3):241-249. Google Scholar Treon SP, Xu L, Yang G. MYD88 L265P somatic mutation in Waldenström's macroglobulinemia. N Engl J Med. 2012; 367(9):826-833. Google Scholar Varettoni M, Zibellini S, Defrancesco I. Pattern of somatic mutations in patients with Waldenström macroglobulinemia or IgM monoclonal gammopathy of undetermined significance. Haematologica. 2017; 102(12):2077-2085. Google Scholar Bazarbachi AH, Avet-Loiseau H, Szalat R. IgM-MM is predominantly a pre-germinal center disorder and has a distinct genomic and transcriptomic signature from WM. Blood. 2021; 138(20):1980-1985. Google Scholar Jäger U, Barcellini W, Broome CM. Diagnosis and treatment of autoimmune hemolytic anemia in adults: recommendations from the First International Consensus Meeting. Blood Rev. 2020; 41:100648. Google Scholar Berentsen S, Barcellini W, D'Sa S. Cold agglutinin disease revisited: a multinational, observational study of 232 patients. Blood. 2020; 136(4):480-488. Google Scholar Berentsen S, Ulvestad E, Langholm R. Primary chronic cold agglutinin disease: a population based clinical study of 86 patients. Haematologica. 2006; 91(4):460-466. Google Scholar Barcellini W, Fattizzo B, Zaninoni A. Clinical heterogeneity and predictors of outcome in primary autoimmune hemolytic anemia: a GIMEMA study of 308 patients. Blood. 2014; 124(19):2930-2936. Google Scholar Röth A, Bommer M, Hüttmann A. Eculizumab in cold agglutinin disease (DECADE): an open-label, prospective, bicentric, nonrandomized phase 2 trial. Blood Adv. 2018; 2(19):2543-2549. Google Scholar Randen U, Trøen G, Tierens A. Primary cold agglutinin-associated lymphoproliferative disease: a B-cell lymphoma of the bone marrow distinct from lymphoplasmacytic lymphoma. Haematologica. 2014; 99(3):497-504. Google Scholar Małecka A, Trøen G, Delabie J. The mutational landscape of cold agglutinin disease: CARD11 and CXCR4 mutations are correlated with lower hemoglobin levels. Am J Hematol. 2021; 96(8):E279-E283. Google Scholar Małecka A, Trøen G, Tierens A. Frequent somatic mutations of KMT2D (MLL2) and CARD11 genes in primary cold agglutinin disease. Br J Haematol. 2018; 183(5):838-842. Google Scholar Malecka A, Delabie J, Ostlie I. Cold agglutinin disease shows highly recurrent gains of chromosome 3, 12 and 18. Blood. 2019; 134(Suppl 1):1488. Google Scholar Berentsen S, Ulvestad E, Gjertsen BT. Rituximab for primary chronic cold agglutinin disease: a prospective study of 37 courses of therapy in 27 patients. Blood. 2004; 103(8):2925-2928. Google Scholar Berentsen S, Randen U, Oksman M. Bendamustine plus ituximab for chronic cold agglutinin disease: results of a Nordic prospective multicenter trial. Blood. 2017; 130(4):537-541. Google Scholar Berentsen S, Randen U, Vågan AM. High response rate and durable remissions following fludarabine and rituximab combination therapy for chronic cold agglutinin disease. Blood. 2010; 116(17):3180-3184. Google Scholar Rossi G, Gramegna D, Paoloni F. Short course of bortezomib in anemic patients with relapsed cold agglutinin disease: a phase 2 prospective GIMEMA study. Blood. 2018; 132(5):547-550. Google Scholar Jalink M, Berentsen S, Castillo JJ. Effect of ibrutinib treatment on hemolytic anemia and acrocyanosis in cold agglutinin disease/cold agglutinin syndrome. Blood. 2021; 138(20):2002-2005. Google Scholar Tomkins O, Berentsen S, Arulogun S, Sekhar M, D'Sa S.. aratumumab for disabling cold agglutinin disease refractory to B-cell directed therapy. Am J Hematol. 2020. Google Scholar Röth A, Barcellini W, D'Sa S. Sutimlimab in cold agglutinin disease. N Engl J Med. 2021; 384(14):1323-1334. Google Scholar Fattizzo B, Michel M, Zaninoni A. Efficacy of recombinant erythropoietin in autoimmune hemolytic anemia: a multicenter international study. Haematologica. 2021; 132(5):622-625. Google Scholar Kolopp-Sarda MN, Nombel A, Miossec P.. Cryoglobulins today: detection and immunologic characteristics of 1,675 positive samples from 13,439 patients obtained over six years. Arthritis Rheumatol. 2019; 71(11):1904-1912. Google Scholar Zhang LL, Cao XX, Shen KN. Clinical characteristics and treatment outcome of type I cryoglobulinemia in Chinese patients: a single-center study of 45 patients. Ann Hematol. 2020; 99(8):1735-1740. Google Scholar Néel A, Perrin F, Decaux O. Long-term outcome of monoclonal (type 1) cryoglobulinemia. Am J Hematol. 2014; 89(2):156-161. Google Scholar Harel S, Mohr M, Jahn I. Clinico-biological characteristics and treatment of type I monoclonal cryoglobulinaemia: a study of 64 cases. Br J Haematol. 2015; 168(5):671-678. Google Scholar Sidana S, Rajkumar SV, Dispenzieri A. Clinical presentation and outcomes of patients with type 1 monoclonal cryoglobulinemia. Am J Hematol. 2017; 92(7):668-673. Google Scholar Terrier B, Karras A, Kahn JE. The spectrum of type I cryoglobulinemia vasculitis: new insights based on 64 cases. Medicine (Baltimore). 2013; 92(2):61-68. Google Scholar Terrier B, Krastinova E, Marie I. Management of noninfectious mixed cryoglobulinemia vasculitis: data from 242 cases included in the CryoVas survey. Blood. 2012; 119(25):5996-6004. Google Scholar Nehme-Schuster H, Korganow AS, Pasquali JL, Martin T.. Rituximab inefficiency during type I cryoglobulinaemia. Rheumatology (Oxford). 2005; 44(3):410-411. Google Scholar Sène D, Ghillani-Dalbin P, Amoura Z, Musset L, Cacoub P.. Rituximab may form a complex with IgMkappa mixed cryoglobulin and induce severe systemic reactions in patients with hepatitis C virus-induced vasculitis. Arthritis Rheum. 2009; 60(12):3848-3855. Google Scholar Wechalekar AD, Lachmann HJ, Goodman HJ, Bradwell A, Hawkins PN, Gillmore JD. AL amyloidosis associated with IgM paraproteinemia: clinical profile and treatment outcome. Blood. 2008; 112(10):4009-4016. Google Scholar Palladini G, Russo P, Bosoni T. AL amyloidosis associated with IgM monoclonal protein: a distinct clinical entity. Clin Lymphoma Myeloma. 2009; 9(1):80-83. Google Scholar Gertz MA, Buadi FK, Hayman SR. IgM amyloidosis: clinical features in therapeutic outcomes. Clin Lymphoma Myeloma Leuk. 2011; 11(1):146-148. Google Scholar Sidana S, Larson DP, Greipp PT. IgM AL amyloidosis: delineating disease biology and outcomes with clinical, genomic and bone marrow morphological features. Leukemia. 2020; 34(5):1373-1382. Google Scholar la Torre A, Reece D, Crump M. Light chain amyloidosis (AL) associated with B cell lymphoma a single center experience. Clin Lymphoma Myeloma Leuk. 2021; 21(12):e946-e959. Google Scholar Sachchithanantham S, Roussel M, Palladini G. European collaborative study defining clinical profile outcomes and novel prognostic criteria in monoclonal immunoglobulin M-related light chain amyloidosis. J Clin Oncol. 2016; 34(17):2037-2045. Google Scholar Terrier B, Jaccard A, Harousseau JL. The clinical spectrum of IgM-related amyloidosis: a French nationwide retrospective study of 72 patients. Medicine (Baltimore). 2008; 87(2):99-109. Google Scholar Kastritis E, Morel P, Duhamel A. A revised international prognostic score system for Waldenström's macroglobulinemia. Leukemia. 2019; 33(11):2654-2661. Google Scholar Venner CP, Lane T, Foard D. Cyclophosphamide, bortezomib, and dexamethasone therapy in AL amyloidosis is associated with high clonal response rates and prolonged progression-free survival. Blood. 2012; 119(19):4387-4390. Google Scholar Sidiqi MH, Buadi FK, Dispenzieri A. Autologous stem cell transplant for IgM-associated amyloid light-chain amyloidosis. Biol Blood Marrow Transplant. 2019; 25(3):e108-e111. Google Scholar Manwani R, Sachchithanantham S, Mahmood S. Treatment of IgM-associated immunoglobulin light-chain amyloidosis with rituximab-bendamustine. Blood. 2018; 132(7):761-764. Google Scholar Palladini G, Foli A, Russo P. Treatment of IgM-associated AL amyloidosis with the combination of rituximab, bortezomib, and dexamethasone. Clin Lymphoma Myeloma Leuk. 2011; 11(1):143-145. Google Scholar Pika T, Hegenbart U, Flodrova P, Maier B, Kimmich C, Schönland SO. First report of ibrutinib in IgM-related amyloidosis: few responses, poor tolerability, and short survival. Blood. 2018; 131(3):368-371. Google Scholar Kastritis E, Palladini G, Minnema MC. Daratumumab-based treatment for immunoglobulin light-chain amyloidosis. N Engl J Med. 2021; 385(1):46-58. Google Scholar Treon SP, Hanzis CA, Ioakimidis LI. Clinical characteristics and treatment outcome of disease-related peripheral neuropathy in Waldenstrom's macroglobulinemia (WM). J Clin Oncol. 2010; 28(15_suppl):8114. Google Scholar Tomkins O, Lindsay J, Keddie S. Neuropathy with IgM gammopathy: incidence, characteristics and management, a Rory Morrison W.M.U.K Registry analysis. Blood. 2020; 136(Suppl 1):1-2. Google Scholar Minnema MC, Kimby E, D'Sa S. Guideline for the diagnosis, treatment and response criteria for Bing-Neel syndrome. Haematologica. 2017; 102(1):43-51. Google Scholar D'Sa S, Kersten MJ, Castillo JJ. Investigation and management of IgM and Waldenström-associated peripheral neuropathies: recommendations from the IWWM-8 consensus panel. Br J Haematol. 2017; 176(5):728-742. Google Scholar Hänggi P, Aliu B, Martin K, Herrendorff R, Steck AJ. Decrease in serum anti-MAG autoantibodies is associated with therapy response in patients with anti-MAG neuropathy: retrospective study. Neurol Neuroimmunol Neuroinflamm. 2022; 9(1):e1109. Google Scholar Lunn MP, Nobile-Orazio E.. Immunotherapy for IgM anti-myelin-associated glycoprotein paraprotein-associated peripheral neuropathies. Cochrane Database Syst Rev. 2016; 10(10):CD002827. Google Scholar Svahn J, Petiot P, Antoine JC. Anti-MAG antibodies in 202 patients: clinicopathological and therapeutic features. J Neurol Neurosurg Psychiatry. 2018; 89(5):499-505. Google Scholar Dalakas MC. Clinical benefits and immunopathological correlates of intravenous immune globulin in the treatment of inflammatory myopathies. Clin Exp Immunol. 1996; 104(Suppl 1):55-60. Google Scholar Nobile-Orazio E, Meucci N, Baldini L, Di Troia A, Scarlato G.. Long-term prognosis of neuropathy associated with anti-MAG IgM M-proteins and its relationship to immune therapies. Brain. 2000; 123(Pt 4):710-717. Google Scholar Le Cann M, Bouhour F, Viala K. CANOMAD: a neurological monoclonal gammopathy of clinical significance that benefits from B-cell-targeted therapies. Blood. 2020; 136(21):2428-2436. Google Scholar Amaador K, Wieske L, Koel-Simmelink MJA. Serum neurofilament light chain, contactin-1 and complement activation in anti-MAG IgM paraprotein-related peripheral neuropathy. J Neurol. 2022; 269(7):3700-3705. Google Scholar Minnema MC, Vos J, Eftimov F, Vrancken A.. P-034: MAGNAZ trial - a prospective phase II study in patients with monoclonal gammopathy of unknown significance (MGUS) and anti-myelin associated glycoprotein (MAG) neuropathy and zanubrutinib treatment. Clin Lymphoma Myeloma Leuk. 2021; 21(Suppl 2):S57. Google Scholar Simon A, Asli B, Braun-Falco M. Schnitzler's syndrome: diagnosis, treatment, and follow-up. Allergy. 2013; 68(5):562-568. Google Scholar Jain T, Offord CP, Kyle RA, Dingli D.. Schnitzler syndrome: an under-diagnosed clinical entity. Haematologica. 2013; 98(10):1581-1585. Google Scholar de Koning HD. Schnitzler's syndrome: lessons from 281 cases. Clin Transl Allergy. 2014; 4:41. Google Scholar Rowczenio DM, Pathak S, Arostegui JI. Molecular genetic investigation, clinical features, and response to treatment in 21 patients with Schnitzler syndrome. Blood. 2018; 131(9):974-981. Google Scholar Lipsker D. The Schnitzler syndrome. Orphanet J Rare Dis. 2010; 5:38. Google Scholar Pathak S, Rowczenio DM, Owen RG. Exploratory study of MYD88 L265P, rare NLRP3 variants, and clonal hematopoiesis prevalence in patients with Schnitzler syndrome. Arthritis Rheumatol. 2019; 71(12):2121-2125. Google Scholar Néel A, Henry B, Barbarot S. Long-term effectiveness and safety of interleukin-1 receptor antagonist (anakinra) in Schnitzler's syndrome: a French multicenter study. Autoimmun Rev. 2014; 13(10):1035-1041. Google Scholar Betrains A, Staels F, Vanderschueren S.. Efficacy and safety of canakinumab treatment in Schnitzler syndrome: a systematic literature review. Semin Arthritis Rheum. 2020; 50(4):636-642. Google Scholar Krause K, Weller K, Stefaniak R. Efficacy and safety of the interleukin-1 antagonist rilonacept in Schnitzler syndrome: an open-label study. Allergy. 2012; 67(7):943-950. Google Scholar Krause K, Feist E, Fiene M, Kallinich T, Maurer M.. Complete remission in 3 of 3 anti-IL-6-treated patients with Schnitzler syndrome. J Allergy Clin Immunol. 2012; 129(3):848-850. Google Scholar Swiecicki PL, Hegerova LT, Gertz MA. Cold agglutinin disease. Blood. 2013; 122(7):1114-1121. Google Scholar Allain JS, Thonier F, Pihan M. IGHV segment utilization in immunoglobulin gene rearrangement differentiates patients with anti-myelin-associated glycoprotein neuropathy from others immunoglobulin M-gammopathies. Haematologica. 2018; 103(5):e207-e210. Google Scholar Vos JM, Notermans NC, D'Sa S. High prevalence of the MYD88 L265P mutation in IgM anti-MAG paraprotein-associated peripheral neuropathy. J Neurol Neurosurg Psychiatry. 2018; 89(9):1007-1009. Google Scholar Kyle RA, Benson J, Larson D. IgM monoclonal gammopathy of undetermined significance and smoldering Waldenström's macroglobulinemia. Clin Lymphoma Myeloma. 2009; 9(1):17-18. Google Scholar Kyle RA, Benson JT, Larson DR. Progression in smoldering Waldenstrom macroglobulinemia: long-term results. Blood. 2012; 119(19):4462-4466. Google Scholar Khwaja J, Patel AS, Arulogun SO. IgM paraprotein-associated type 1 cryoglobulinaemia: clinical characteristics and outcomes. Blood. 2021; 138(Suppl 1):4503-4503. Google Scholar Nobile-Orazio E, Barbieri S, Baldini L. Peripheral neuropathy in monoclonal gammopathy of undetermined significance: prevalence and immunopathogenetic studies. Acta Neurol Scand. 1992; 85(6):383-390. Google Scholar
|
|
Scooped by
Gilbert C FAURE
August 17, 2022 3:45 AM
|
The US veteran population has a high proportion of chronic lymphocytic leukemia (CLL) risk factors. Using the Veterans Health Administration (VHA) population, we conducted a retrospective chart rev...
|
|
Scooped by
Gilbert C FAURE
July 20, 2022 1:30 PM
|
|
|
Scooped by
Gilbert C FAURE
July 4, 2022 6:35 AM
|
The classification of myeloid neoplasms and acute leukemias was last updated in 2016 within a collaboration between the World Health Organization (WHO), the Society for Hematopathology, and the European Association for Haematopathology.
|
|
Scooped by
Gilbert C FAURE
June 20, 2022 10:44 AM
|
The origins of our blood may not be quite what we thought. Using cellular "barcoding" in mice, a groundbreaking study finds that blood cells originate not from one type of mother cell, but two, with potential implications for blood cancers, bone marrow transplant, and immunology. Fernando Camargo, Ph.D., of the Stem Cell Program at Boston Children's Hospital led the study, published in Nature on June 15.
|
|
Scooped by
Gilbert C FAURE
April 28, 2022 3:11 AM
|
Large granular lymphocyte leukemia (LGLL) is a chronic disease of either mature phenotype cytotoxic CD3+ T lymphocytes or CD3- NK cells. LGLL diagnosis is hampered by the fact that reactive persistent clonal LGL expansions may fulfill the current criteria for LGLL diagnoses.
|
|
Scooped by
Gilbert C FAURE
April 20, 2022 3:16 AM
|
An otherwise healthy 46-year-old woman presents with sharp, severe, diffuse abdominal pain that awakened her from sleep. Do you know what's wrong?
|
|
Scooped by
Gilbert C FAURE
March 22, 2022 6:12 AM
|
Plasma cell neoplasms (including multiple myeloma) treatment include observation, chemotherapy, radiation, stem cell rescue, targeted, and supportive therapies. Corticosteroids and immunomodulatory drugs may be used. Get detailed treatment information in this summary for clinicians.
|
|
Scooped by
Gilbert C FAURE
March 1, 2022 11:51 AM
|
Over the last few years, treatment principles have been changed towards more targeted therapy for many B-cell lymphoma subtypes and in chronic lymphocytic leukemia (CLL). Immunotherapeutic modalities, namely monoclonal antibodies (mAbs), bispecific antibodies (bsAbs), antibody-drug conjugates...
|
|
Scooped by
Gilbert C FAURE
February 21, 2022 3:50 AM
|
Immune-based therapeutic strategies have drastically changed the landscape of hematological disorders, as they have introduced the concept of boosting immune responses against tumor cells. Anti-CD20 monoclonal antibodies have been the first form of immunotherapy successfully applied in the...
|
|
|
Scooped by
Gilbert C FAURE
October 19, 2022 4:34 AM
|
Diagnosis and classification of tumors is increasingly dependent on biomarkers. RNA expression profiling using next-generation sequencing (NGS) provides reliable and reproducible information on the biology of cancer.
|
|
Scooped by
Gilbert C FAURE
October 5, 2022 12:16 PM
|
Multiple myeloma (MM) is the second most common blood cancer. Treatments for MM include corticosteroids, alkylating agents, anthracyclines, proteasome inhibitors, immunomodulatory drugs, histone deacetylase inhibitors and monoclonal antibodies.
|
|
Scooped by
Gilbert C FAURE
September 27, 2022 3:45 AM
|
Follicular lymphoma (FL) originates from germinal center B cells, is the most prevalent form of indolent non-Hodgkin’s lymphoma. Upfront management is based on stage, grade, and disease burden. Radiotherapy may be curative in limited disease while ...
|
|
Scooped by
Gilbert C FAURE
September 22, 2022 11:03 AM
|
Cheng H, Cheng T. ‘Waterloo’: when normal blood cells meet leukemia. Curr Opin Hematol. 2016;23(4):304–310.View this article via: CrossRef PubMed Google Scholar Hoggatt J, et al. Hematopoietic stem cell niche in health and disease. Annu Rev Pathol.
|
|
Scooped by
Gilbert C FAURE
August 2, 2022 1:55 PM
|
Check out this study that contributes to identify the patients with B-ALL at risk of CD19-targeted therapy failure.
|
|
Scooped by
Gilbert C FAURE
July 13, 2022 10:10 AM
|
Background: Machine learning (ML) and deep learning (DL) methods have recently garnered a great deal of attention in the field of cancer resear.
|
|
Scooped by
Gilbert C FAURE
June 26, 2022 11:20 AM
|
Unconventional T cells and innate lymphoid cells (ILCs) make up a heterogeneous set of cells that characteristically show prompt responses toward specific antigens.Unconventional T cells recognize non-peptide antigens, which are bound and presented by diverse non-polymorphic antigen-presenting mole...
|
|
Scooped by
Gilbert C FAURE
April 28, 2022 3:37 AM
|
PURPOSE Most cases of pediatric acute leukemia occur in low- and middle-income countries, where health centers lack the tools required for accurate diagnosis and disease classification. Recent research shows the robustness of using unbiased short-read RNA sequencing to classify genomic subtypes of acute leukemia. Compared with short-read sequencing, nanopore sequencing has low capital and consumable costs, making it suitable for use in locations with limited health infrastructure. MATERIALS AND METHODS We show the feasibility of nanopore mRNA sequencing on 134 cryopreserved acute leukemia specimens (26 acute myeloid leukemia [AML], 73 B-lineage acute lymphoblastic leukemia [B-ALL], 34 T-lineage acute lymphoblastic leukemia, and one acute undifferentiated leukemia). Using multiple library preparation approaches, we generated long-read transcripts for each sample. We developed a novel composite classification approach to predict acute leukemia lineage and major B-ALL and AML molecular subtypes directly from gene expression profiles. RESULTS We demonstrate accurate classification of acute leukemia samples into AML, B-ALL, or T-lineage acute lymphoblastic leukemia (96.2% of cases are classifiable with a probability of > 0.8, with 100% accuracy) and further classification into clinically actionable genomic subtypes using shallow RNA nanopore sequencing, with 96.2% accuracy for major AML subtypes and 94.1% accuracy for major B-lineage acute lymphoblastic leukemia subtypes. CONCLUSION Transcriptional profiling of acute leukemia samples using nanopore technology for diagnostic classification is feasible and accurate, which has the potential to improve the accuracy of cancer diagnosis in low-resource settings.
|
|
Scooped by
Gilbert C FAURE
April 20, 2022 4:32 AM
|
Using single-cell RNA sequencing, Ye et al. demonstrate that genetic diversity within
tumors, potential interactions between malignant and tumor-infiltrating cells, and
viral infections may all contribute to the marked disease heterogeneity in DLBCL.
|
|
Scooped by
Gilbert C FAURE
March 27, 2022 4:55 AM
|
Diabetes mellitus is a complex disease that leads to long-term damage to various organ systems. Among the numerous cardiovascular disease-related complications, thrombotic events frequently occur i...
|
|
Scooped by
Gilbert C FAURE
March 17, 2022 5:40 AM
|
|
|
Scooped by
Gilbert C FAURE
February 22, 2022 4:40 AM
|
Both qualitative and quantitative platelet abnormalities are common in patients with coronavirus disease 2019 (COVID-19) and they correlate with clinical severity and mortality. Activated platelets contribute to the prothrombotic state in COVID-19 patients. Several groups have shown immune-mediated activation of platelets in critically ill COVID-19 patients. Vaccine-induced immune thrombotic thrombocytopenia is an autoimmune condition characterized by thrombocytopenia and life-threatening thrombotic events in the arterial and venous circulation. Although the initial trigger has yet to be determined, activation of platelets by immune complexes through Fc gamma RIIA results in platelet consumption and thrombosis. A better understanding of platelet activation in COVID-19 as well as in vaccine-induced thrombotic complications will have therapeutic implications. In this review, we focused on the role of immune-mediated platelet activation in thrombotic complications during COVID-19 infection and vaccine-induced immune thrombotic thrombocytopenia.
|