Your new post is loading...

|
Rescooped by
Dr. Stefan Gruenwald
from Virus World
February 21, 2023 3:18 PM
|
A 53-year-old man in Germany has become the third person with HIV to be declared cleared of the virus after a procedure that replaced his bone marrow cells with HIV-resistant stem cells from a donor. For years, antiretroviral therapy (ART) has been given to people with HIV with the aim of lowering the virus to almost undetectable levels and preventing it from being transmitted to other people. But the immune system keeps the virus locked up in reservoirs in the body, and if an individual stops taking ART the virus can begin replicating and spreading. A true cure would eliminate this reservoir, and this is what seems to have happened for the latest patient, whose name has not been released. The man, who is being referred to as the ‘Düsseldorf patient’, stopped taking ART in 2018 and has remained HIV-free since. But the risks associated with the procedure mean it is unlikely to be widely used in its current form. The stem-cell technique involved was first used to treat Timothy Ray Brown, often referred to as the Berlin patient. In 2007, he had a bone marrow transplant, in which those cells were destroyed and replaced with stem cells from a healthy donor, to treat acute myeloid leukemia. The team treating Brown selected a donor with a genetic mutation called CCR5Δ32/Δ32, which prevents the CCR5 cell-surface protein from being expressed on the cell surface. HIV uses that protein to enter immune cells, so the mutation makes the cells effectively resistant to the virus. After the procedure, Brown was able to stop taking ART and remained HIV-free until his death in 2020. In 2019, researchers revealed that the same procedure seemed to have cured the London patient, Adam Castillejo. And, in 2022, scientists announced that they thought a New York patient who had remained HIV-free for 14 months might also be cured, although researchers cautioned that it was too early to be certain. Ravindra Gupta, a microbiologist at the University of Cambridge, UK, who led the team that treated Castillejo, says the latest study “cements the fact that CCR5 is the most tractable target for achieving a cure right now”. Low virus levels The Düsseldorf patient had extremely low levels of HIV, thanks to ART, when he was diagnosed with acute myeloid leukaemia. In 2013, a team led by virologist Björn-Erik Jensen at Düsseldorf University Hospital in Germany destroyed the patient’s cancerous bone marrow cells and replaced them with stem cells from a donor with the CCR5Δ32/Δ32 mutation1. Over the next five years, Jensen’s team took tissue and blood samples from the patient. In the years after the transplant, the scientists continued to find immune cells that specifically reacted to HIV, which suggested that a reservoir remained somewhere in the man’s body. It’s not clear, Jensen says, whether these immune cells had targeted active virus particles or a “graveyard” of viral remnants. They also found HIV DNA and RNA in the patient’s body, but these never seemed to replicate. In an effort to understand more about how the transplant worked, the team ran further tests, which included transplanting the patient’s immune cells into mice engineered to have human-like immune systems. The virus failed to replicate in the mice, suggesting that it was nonfunctional. The final test was for the patient to stop taking ART. “It shows it’s not impossible — it’s just very difficult — to remove HIV from the body,” Jensen says. The patient who received the treatment said in a statement that the bone marrow transplant had been a “very rocky road”, adding that he planned to devote some of his life to supporting research fundraising. Timothy Henrich, an infectious-disease researcher at the University of California, San Francisco, says the study is very thorough. That several patients have been successfully treated with a combination of ART and HIV-resistant donor cells makes the chances of achieving an HIV cure in these individuals very high. Gupta agrees, although he adds that in some cases the virus mutates inside a person and finds other ways to enter their cells. It’s also unclear, he says, whether the chemotherapy that the people received for their cancer before their bone marrow transplants might have helped to eliminate HIV by preventing infected cells from dividing. But it’s unlikely that bone-marrow replacement will be rolled out to people who don’t have leukemia because of the high risk associated with the procedure, particularly the chance that an individual will reject a donor’s marrow. Several teams are testing the potential to use stem cells taken from a person’s own body and then genetically modified to have the CCR5Δ32/Δ32 mutation2,3, which would eliminate the need for donor cells. Jensen says that his team has performed transplants for several other people affected by both HIV and cancer using stem cells from donors with a CCR5Δ32/Δ32 mutation, but that it is too early to say whether those individuals are virus-free. His team plans to study whether, if a person has a larger reservoir of HIV at the time of receiving a transplant, this affects how well the immune system recovers and eliminates any remaining viruses from the body. Case report published in Nature Medicine (Feb. 20, 2023): https://doi.org/10.1038/s41591-023-02213-x
Via Juan Lama
|
|
Scooped by
Dr. Stefan Gruenwald
December 24, 2022 1:56 PM
|
Cat genes reveal how invention of agriculture bonded cats with people in ancient Mesopotamia, leading to worldwide feline migration with humans. Nearly 10,000 years ago, humans settling in the Fertile Crescent, the areas of the Middle East surrounding the Tigris and Euphrates rivers, made the first switch from hunter-gatherers to farmers. They developed close bonds with the rodent-eating cats that conveniently served as ancient pest-control in society's first civilizations. A new study at the University of Missouri found this lifestyle transition for humans was the catalyst that sparked the world's first domestication of cats, and as humans began to travel the world, they brought their new feline friends along with them. Leslie A. Lyons, a feline geneticist and Gilbreath-McLorn endowed professor of comparative medicine in the MU College of Veterinary Medicine, collected and analyzed DNA from cats in and around the Fertile Crescent area, as well as throughout Europe, Asia and Africa, comparing nearly 200 different genetic markers. "One of the DNA main markers we studied were microsatellites, which mutate very quickly and give us clues about recent cat populations and breed developments over the past few hundred years," Lyons said. "Another key DNA marker we examined were single nucleotide polymorphisms, which are single-based changes all throughout the genome that give us clues about their ancient history several thousands of years ago. By studying and comparing both markers, we can start to piece together the evolutionary story of cats." Lyons added that while horses and cattle have seen various domestication events caused by humans in different parts of the world at various times, her analysis of feline genetics in the study strongly supports the theory that cats were likely first domesticated only in the Fertile Crescent before migrating with humans all over the world. After feline genes are passed down to kittens throughout generations, the genetic makeup of cats in western Europe, for example, is now far different from cats in southeast Asia, a process known as 'isolation by distance.' "We can actually refer to cats as semi-domesticated, because if we turned them loose into the wild, they would likely still hunt vermin and be able to survive and mate on their own due to their natural behaviors," Lyons said. "Unlike dogs and other domesticated animals, we haven't really changed the behaviors of cats that much during the domestication process, so cats once again prove to be a special animal." Lyons, who has researched feline genetics for more than 30 years, said studies like this also support her broader research goal of using cats as a biomedical model to study genetic diseases that impact both cats and people, such as polycystic kidney disease, blindness and dwarfism. "Comparative genetics and precision medicine play key roles in the 'One Health' concept, which means anything we can do to study the causes of genetic diseases in cats or how to treat their ailments can be useful for one day treating humans with the same diseases," Lyons said. "I am building genetic tools, genetic resources that ultimately help improve cat health. When building these tools, it is important to get a representative sample and understand the genetic diversity of cats worldwide so that our genetic toolbox can be useful to help cats all over the globe, not just in one specific region."
|
|
Rescooped by
Dr. Stefan Gruenwald
from Virus World
December 8, 2022 11:32 PM
|
An mRNA vaccine has been found to induce antibody responses against all 20 known subtypes of influenza A and B in mice and ferrets. An experimental vaccine has generated antibody responses against all 20 known strains of influenza A and B in animal tests, raising hopes for developing a universal flu vaccine. Influenza viruses are constantly evolving, making them a moving target for vaccine developers. The annual flu vaccines available now are tailored to give immunity against specific strains predicted to circulate each year. However, researchers sometimes get the prediction wrong, meaning the vaccine is less effective than it could be in those years. Some researchers think annual flu jabs could be replaced by a universal flu vaccine that is effective against all flu strains. Researchers have tried to achieve this by making vaccines containing protein fragments that are common to several influenza strains, but no universal vaccine has yet gained approval for wider use. Now, Scott Hensley at the University of Pennsylvania and his colleagues have created a vaccine based on mRNA molecules – the same approach that was pioneered by the Pfizer/BioNTech and Moderna covid-19 vaccines. mRNA contains genetic codes for making proteins, just like DNA. The vaccine contains mRNA molecules encoding fragments of proteins found in all 20 known strains of influenza A and B – the viruses that cause seasonal outbreaks each year. The strains have different versions of two proteins on their surface, haemagglutinin (H) and neuraminidase (N), which are targeted by immune responses. But even within one strain, such as H1N1, there can be slight variations in these proteins, so the version in the universal vaccine will not exactly match every possible variant. In tests in mice, the team found that the animals generated antibodies specific to all 20 strains of the flu virus, and these antibodies remained at a stable level for up to four months. In another test, the team gave mice the universal flu vaccine or a dummy vaccine containing code for a non-flu protein. A month later, they infected them with either one of two variants of the H1N1 flu virus, one with an H1 protein that was very similar to the version of the protein in the vaccine, and one with a more distinct version. All the mice given the flu vaccine survived exposure to the virus with the more similar protein and 80 per cent survived being infected with the more distinct variant. All of the mice given the dummy vaccine died around a week after infection with either variant. Another group of mice were given an mRNA vaccine targeted only to the precise flu strain they were exposed to, and all of this group survived over the same time period. This suggests the universal flu vaccine would offer less protection against new variants of the 20 flu strains than an annual vaccine matched to new forms of the virus, says Albert Osterhaus at the University of Veterinary Medicine Hannover in Germany, who wasn’t involved in the study. The researchers also tested the universal vaccine in ferrets with similar results. “The mouse and ferret models for influenza are as good as animal models get. The animal data are promising and thus a good indication of what will happen in humans,” says Peter Palese at the Icahn School of Medicine at Mount Sinai in New York. A key benefit of mRNA vaccines is that they can easily be scaled up compared with other approaches which rely on growing influenza viruses in chicken eggs or in the lab, says Palese. “For generating a basic immunity against epidemic or pandemic influenza virus strains in the future, this strategy could offer an option if longevity [of immunity] in humans is confirmed,” says Osterhaus. “Definitely these animal data are promising and merit further exploration in clinical studies. Given previous studies with candidate universal flu vaccines in human trials, it is hard to predict what the clinical data will bring,” says Osterhaus. “This 20-HA mRNA vaccine was tested in ferret animals, which is highly significant and may hold promise for protecting against future emerging flu strains against severe disease in humans,” says Sang-Moo Kang at Georgia State University. Study cited published in Science (Nov. 24): https://doi.org/10.1126/science.abm0271
Via Juan Lama
|
|
Rescooped by
Dr. Stefan Gruenwald
from Virus World
December 1, 2022 1:37 AM
|
New Research Finds That Viruses May Have “Eyes and Ears” on Us The newly-found, widespread ability of some viruses to monitor their environment could have implications for antiviral drug development. New research indicates that viruses are using information from their environment to “decide” when to sit tight inside their hosts and when to multiply and burst out, killing the host cell. The work has important implications for antiviral drug development. Led by the University of Maryland Baltimore County (UMBC), the study was recently published in Frontiers in Microbiology. A virus’s ability to sense its environment, including elements produced by its host, adds “another layer of complexity to the viral-host interaction,” says Ivan Erill. He is senior author on the new paper and professor of biological sciences at UMBC. Right now, viruses are taking advantage of that ability to their benefit. But he says that in the future, “we could exploit it to their detriment.” Not a coincidence The new study focused on bacteriophages, which are often referred to simply as “phages.” They are viruses that infect bacteria. In the study, the phages analyzed can only infect their hosts when the bacterial cells have special appendages, called pili and flagella, that help the bacteria move and mate. The bacteria produce a protein called CtrA that controls when they generate these appendages. The research revealed that many appendage-dependent phages have patterns in their DNA where the CtrA protein can attach, called binding sites. Erill says that a phage having a binding site for a protein produced by its host is unusual. Even more surprising, Erill and the paper’s first author Elia Mascolo, a Ph.D. student in Erill’s lab, discovered through detailed genomic analysis that these binding sites were not unique to a single phage, or even a single group of phages. Many different types of phages had CtrA binding sites—but they all required their hosts to have pili and/or flagella to infect them. They decided that it couldn’t be a coincidence. The ability to monitor CtrA levels “has been invented multiple times throughout evolution by different phages that infect different bacteria,” Erill says. When distantly related species exhibit a similar trait, it’s called convergent evolution—and it indicates that the trait is definitely useful. Timing is everything Another wrinkle in the story: The first phage in which the scientists identified CtrA binding sites infects a particular group of bacteria called Caulobacterales. Caulobacterales are an especially well-studied group of bacteria, because they exist in two forms: a “swarmer” form that swims around freely, and a “stalked” form that attaches to a surface. The swarmers have pili/flagella, and the stalks do not. In these bacteria, CtrA also regulates the cell cycle, determining whether a cell will divide evenly into two more of the same cell type, or divide asymmetrically to produce one swarmer and one stalk cell. Since the phages can only infect swarmer cells, it’s in their best interest only to burst out of their host when there are many swarmer cells available to infect. Generally, Caulobacterales live in nutrient-poor environments, and they are very spread out. “But when they find a good pocket of microhabitat, they become stalked cells and proliferate,” Erill says, eventually producing large quantities of swarmer cells. So, “We hypothesize the phages are monitoring CtrA levels, which go up and down during the life cycle of the cells, to figure out when the swarmer cell is becoming a stalk cell and becoming a factory of swarmers,” Erill says, “and at that point, they burst the cell, because there are going to be many swarmers nearby to infect.” Listening in Unfortunately, the method to prove this hypothesis is extremely difficult and labor-intensive, so that wasn’t part of this latest paper—although Erill and colleagues hope to tackle that question in the future. However, the research team sees no other plausible explanation for the proliferation of CtrA binding sites on so many different phages, all of which require pili/flagella to infect their hosts. Even more interesting, they note, are the implications for viruses that infect other organisms—even humans. “Everything that we know about phages, every single evolutionary strategy they have developed, has been shown to translate to viruses that infect plants and animals,” he says. “It’s almost a given. So if phages are listening in on their hosts, the viruses that affect humans are bound to be doing the same.” There are a few other documented examples of phages monitoring their environment in interesting ways, but none include so many different phages employing the same strategy against so many bacterial hosts. This new research is the “first broad scope demonstration that phages are listening in on what’s going on in the cell, in this case, in terms of cell development,” Erill says. But more examples are on the way, he predicts. Already, members of his lab have started looking for receptors for other bacterial regulatory molecules in phages, he says—and they’re finding them. New therapeutic avenues The key takeaway from this research is that “the virus is using cellular intel to make decisions,” Erill says, “and if it’s happening in bacteria, it’s almost certainly happening in plants and animals, because if it’s an evolutionary strategy that makes sense, evolution will discover it and exploit it.” For example, an animal virus might want to know what kind of tissue it is in, or how robust the host’s immune response is to its infection in order to optimize its strategy for survival and replication. While it might be disturbing to think about all the information viruses could gather and possibly use to make us sicker, these discoveries also open up opportunities for new therapies. “If you are developing an antiviral drug, and you know the virus is listening in on a particular signal, then maybe you can fool the virus,” Erill says. That’s several steps away, however. For now, “We are just starting to realize how actively viruses have eyes on us—how they are monitoring what’s going on around them and making decisions based on that,” Erill says. “It’s fascinating.” Research Cited Published in Frontiers in Microbiology (August 17, 2022): https://doi.org/10.3389/fmicb.2022.918015
Via Juan Lama
|
|
Rescooped by
Dr. Stefan Gruenwald
from Virus World
November 16, 2022 9:24 PM
|
Monkeypox has infected more than 77,000 people in more than 100 countries worldwide, and—similar to COVID-19—mutations have enabled the virus to grow stronger and smarter, evading antiviral drugs and vaccines in its mission to infect more people. Now, a team of researchers at the University of Missouri have identified the specific mutations in the monkeypox virus that contribute to its continued infectiousness. The findings could lead to several outcomes: modified versions of existing drugs used to treat people suffering from monkeypox or the development of new drugs that account for the current mutations to increase their effectiveness at reducing symptoms and the spread of the virus. Kamlendra Singh, a professor in the MU College of Veterinary Medicine and Christopher S. Bond Life Sciences Center principal investigator, collaborated with Shrikesh Sachdev, Shree Lekha Kandasamy and Hickman High School student Saathvik Kannan, to analyze the DNA sequences of more 200 strains of monkeypox virus spanning multiple decades, from 1965, when the virus first started spreading, to outbreaks in the early 2000s and again in 2022. "By doing a temporal analysis, we were able to see how the virus has evolved over time, and a key finding was the virus is now accumulating mutations specifically where drugs and antibodies from vaccines are supposed to bind," Sachdev said. "So, the virus is getting smarter, it is able to avoid being targeted by drugs or antibodies from our body's immune response and continue to spread to more people." Needles in a haystack Singh has been studying virology and DNA genome replication for nearly 30 years. He said the homology, or structure, of the monkeypox virus is very similar to the vaccinia virus, which has been used as a vaccine to treat smallpox. This enabled Singh and his collaborators to create an accurate, 3D computer model of the monkeypox virus proteins and identify both where the specific mutations are located and what their functions are in contributing to the virus becoming so infectious recently. "Our focus is on looking at the specific genes involved in copying the virus genome, and monkeypox is a huge virus with approximately 200,000 DNA bases in the genome," Singh said. "The DNA genome for monkeypox is converted into nearly 200 proteins, so it comes with all the 'armor' it needs to replicate, divide and continue to infect others. Viruses will make billions of copies of itself and only the fittest will survive, as the mutations help them adapt and continue to spread." Kannan and Kandasamy examined five specific proteins while analyzing the monkeypox virus strains: DNA polymerase, DNA helicase, bridging protein A22R, DNA glycosylase and G9R. "When they sent me the data, I saw that the mutations were occurring at critical points impacting DNA genome binding, as well as where drugs and vaccine-induced antibodies are supposed to bind," Singh said. "These factors are surely contributing to the virus' increased infectivity. This work is important because the first step toward solving a problem is identifying where the problem is specifically occurring in the first place, and it is a team effort." The evolution of viruses Researchers continue to question how the monkeypox virus has evolved over time. The efficacy of current CDC-approved drugs to treat monkeypox have been suboptimal, likely because they were originally developed to treat HIV and herpes but have since received emergency use authorization in an attempt to control the recent monkeypox outbreak. "One hypothesis is when patients were being treated for HIV and herpes with these drugs, they may have also been infected with monkeypox without knowing, and the monkeypox virus got smarter and mutated to evade the drugs," Singh said. "Another hypothesis is the monkeypox virus may be hijacking proteins we have in our bodies and using them to become more infectious and pathogenic." Singh and Kannan have been collaborating since the COVID-19 pandemic began in 2020, identifying the specific mutations causing COVID-19 variants, including Delta and Omicron. Kannan was recently recognized by the United Nations for supporting their 'Sustainable Development Goals,' which help tackle the world's greatest challenges. "I could not have done this research without my team members, and our efforts have helped scientists and drug developers assist with these virus outbreaks, so it is rewarding to be a part of it," Singh said. "Mutations in the monkeypox virus replication complex: Potential contributing factors to the 2022 outbreak" was recently published in Journal of Autoimmunity. Co-authors on the study include Shrikesh Sachdev, Athreya Reddy, Shree Lekha Kandasamy, Siddappa Byrareddy, Saathvik Kannan and Christian Lorson. Research cited published in Journal of Autoimmunity (Dec. 22, 2022): https://doi.org/10.1016/j.jaut.2022.102928
Via Juan Lama
|
|
Scooped by
Dr. Stefan Gruenwald
October 24, 2022 1:08 PM
|
Methane traps 30 times more heat than carbon dioxide and is estimated to account for about 30 percent of human-driven global warming. The gas is emitted naturally through geological processes and by methane-generating archaea; however, industrial processes are releasing stored methane back into the atmosphere in worrying quantities. Last year, a team led by Jill Banfield discovered DNA structures within a methane-consuming microbe called Methanoperedens that appear to supercharge the organism’s metabolic rate. They named the genetic elements “Borgs” because the DNA within them contains genes assimilated from many organisms. In a study published today in Nature, the researchers describe the curious collection of genes within Borgs and begin to investigate the role these DNA packages play in environmental processes, such as carbon cycling. Methanoperedens are a type of archaea (unicellular organisms that resemble bacteria but represent a distinct branch of life) that break down methane (CH4) in soils, groundwater, and the atmosphere to support cellular metabolism. Methanoperedens and other methane-consuming microbes live in diverse ecosystems around the world but are believed to be less common than microbes that use photosynthesis, oxygen, or fermentation for energy. Yet they play an outsized role in Earth system processes by removing methane – the most potent greenhouse gas – from the atmosphere. Banfield, a faculty scientist at Lawrence Berkeley National Laboratory (Berkeley Lab) and professor of Earth & Planetary Science and Environmental Science, Policy & Management at UC Berkeley, studies how microbial activities shape large-scale environmental processes and how, in turn, environmental fluctuations alter the planet’s microbiomes. As part of this work, she and her colleagues regularly sample microbes in different habitats to see what interesting genes microbes are using for survival, and how these genes might affect global cycles of key elements, such as carbon, nitrogen, and sulfur. The team looks at the genomes within cells as well as the portable packets of DNA known as extra-chromosomal elements (ECEs) that transfer genes between bacteria, archaea, and viruses. These elements allow microbes to quickly gain beneficial genes from their neighbors, including those that are only distantly related. While studying Methanoperedens sampled from seasonal wetland pool soil in California, the scientists found evidence of an entirely new type of ECE. Unlike the circular strands of DNA that make up most plasmids, the most well-known type of extra-chromosomal element, the new ECEs are linear and very long – up to one-third the length of the entire Methanoperedens genome. After analyzing additional samples from underground soil, aquifers, and riverbeds in California and Colorado that contain methane-consuming archaea, the team uncovered a total of 19 distinct ECEs they dubbed Borgs. Using advanced genome analysis tools, the scientists determined that many of the sequences within the Borgs are similar to the methane-metabolizing genes within the actual Methanoperedens genome. Some of the Borgs even encode all the necessary cellular machinery to eat methane on their own, so long as they are inside a cell that can express the genes.
|
|
Rescooped by
Dr. Stefan Gruenwald
from Virus World
October 8, 2022 5:19 PM
|
A recent experiment tests whether the gene-editing technology CRISPR can stop the virus from replicating, which would ultimately wipe out the HIV infection. In July, an HIV-positive man became the first volunteer in a clinical trial aimed at using the CRISPR gene editing technology to snip the HIV virus out of his cells. For an hour, he was hooked up to an IV-bag that pumped the experimental treatment directly into his bloodstream. The one-time infusion is designed to carry the gene-editing tools to the man’s infected cells to clear the virus. Later this month, the volunteer will stop taking the anti-retroviral drugs he’s been on to keep the virus at undetectable levels. Then, investigators will wait 12 weeks to see if the virus rebounds and becomes detectable again. If not, they’ll consider the experiment a success. “What we’re trying to do is return the cell to a near-normal state,” says Daniel Dornbusch, CEO of Excision BioTherapeutics, the San Francisco-based biotech company that’s running the trial. HIV attacks CD4-positive immune cells in the body and hijacks their machinery to make copies of itself. But some HIV-infected cells can go dormant—sometimes for years—and not actively produce new copies of the virus. These so-called reservoirs are a major barrier to curing HIV. “HIV is a tough foe to fight because it’s able to insert itself into our own DNA, and it’s also able to become silent and reactivate at different points in a person’s life,” says Jonathan Li, a physician at Brigham and Women’s Hospital and HIV researcher at Harvard University who’s not involved with the CRISPR trial. Figuring out how to target these reservoirs—and doing it without harming vital CD4 cells—has proven challenging, Li says. While antiretroviral drugs can halt viral replication and clear the virus from the blood, they can’t reach these hide-out reservoirs, so people have to take medication every day for the rest of their lives. But Excision BioTherapeutics is hoping that this CRISPR-based approach will remove HIV for good. CRISPR is being used in several other studies to treat a handful of conditions that arise from various genetic mutations. In those cases, scientists are using CRISPR to edit peoples’ own cells. But for the HIV trial, Excision researchers are turning the gene-editing tool against the virus. The CRISPR infusion contains gene-editing molecules that target two regions in the HIV genome important for viral replication. The virus can only reproduce if it’s fully intact, so CRISPR disrupts that process by cutting out chunks of the genome. In 2019, researchers at Temple University and the University of Nebraska found that using CRISPR to delete those regions eliminated HIV from the genomes of rats and mice. A year later, the Temple group also showed that the approach safely removed viral DNA from macaques with SIV, the monkey version of HIV. That was an important step toward testing the treatment in people, says Kamel Khalili, a professor of microbiology at Temple University who led the work and is a cofounder of Excision Biotherapeutics. “You don’t want to eliminate the viral genome but at the same time cause any disruption in another part of the human genome and then create another set of problems for the patients,” he says. “We had to make sure that we identified a region within HIV that did not overlap with the human genome.” Dornbusch thinks this strategy will spare patients from serious side effects and “off-target” edits—unintentional cuts elsewhere in the genome that could cause problems such as cancer. The regions targeted by the company’s CRISPR therapy are also in a part of the genome that tends to stay the same even when HIV evolves. That’s important because the virus mutates rapidly, and the researchers don’t want a moving target. This isn’t the first time scientists have tried to use gene editing in the hope of curing people with HIV, but other efforts have focused on a protective mutation in a gene called CCR5. In the 1990s, scientists found that people with this naturally occurring mutation didn’t get HIV even when exposed to it. The mutation—known as delta 32—thwarts the virus’s ability to get inside immune cells. In 2009, California-based Sangamo Therapeutics used an older editing technology involving zinc finger nucleases to add that protective mutation into patients’ T cells—an important part of the immune system. Those trials have had only limited success. In 2017, Chinese scientists combined CRISPR with a bone marrow transplant in an attempt to cure a patient with HIV and leukemia. In a typical transplant, donor stem cells are transferred to a recipient to replace their cancerous blood cells. These cells go on to form new, healthy blood cells. To also address the patient’s HIV, researchers edited the donor stem cells with CRISPR to disable CCR5. But after the transplant, only a small percentage of the patient’s bone marrow cells ended up with the desired edit. Then in 2018, Chinese scientist He Jiankui used CRISPR to edit the CCR5 mutation into the genomes of twin baby girls to make them resistant to HIV during their lives. Fraught with ethical violations, the experiment was widely condemned by scientists. He’s research was suspended by the Chinese government, and he served a three-year prison sentence. While the twins were born healthy, only some of their cells were successfully edited, meaning the girls might in fact not be immune to HIV. As of 2022, two people have now been cured of HIV after receiving bone marrow transplants from donors with the CCR5. Known as the Berlin patient and the London patient, both had cancer and received transplants to treat their disease. But these transplants aren’t a viable option for most people—they’re highly risky, and donors with the delta 32 mutation are scarce. But a third person was declared cured of HIV earlier this year after she received a new type of transplant involving umbilical cord blood. The Excision trial will eventually enroll nine participants and test three dosage amounts to determine which is most effective. Investigators will measure each person’s viral load and CD4 count before receiving the therapy and after they stop taking anti-retroviral drugs. The ultimate goal is to get viral loads down to an undetectable level—that is, less than 200 copies of HIV per milliliter of blood. At this level, HIV can’t be passed on through sex anymore. The challenge for Excision will be getting CRISPR to enough cells to bring HIV down to undetectable levels. The company is using an engineered virus to shuttle the gene-editing components to patients’ HIV-infected CD4 cells. But so far, there’s little human data on how well CRISPR works when it’s delivered directly to the body. “It’s possible that you get the virus to such low levels that if a person’s immune system were intact, they might be able to keep the virus at bay such that they don’t have to take antiretroviral therapy anymore,” says Rowena Johnston, vice president and director of research for amfAR, the Foundation for AIDS Research. And even though these drugs are very effective, Johnston says, many people would rather be completely free of the virus. A single CRISPR infusion—if it works—would eliminate the need for daily pills. “People with HIV still live with a lot of stigma and internalized shame,” she says. “I think a cure is something that addresses that much better than lifelong therapy, regardless of how easy that therapy becomes.”
Via Juan Lama
|
|
Scooped by
Dr. Stefan Gruenwald
October 4, 2022 12:40 PM
|
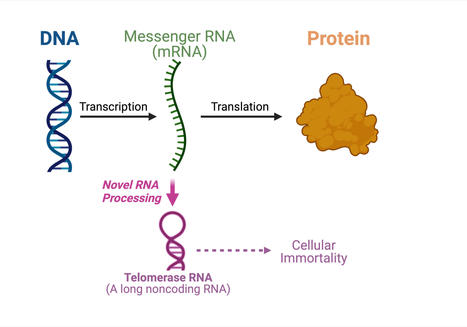
For the very first time, a study led by Julian Chen and his group in Arizona State University's School of Molecular Sciences and the Biodesign Institute's Center for the Mechanism of Evolution, has discovered an unprecedented pathway producing telomerase RNA from a protein-coding messenger RNA (mRNA). The central dogma of molecular biology specifies the order in which genetic information is transferred from DNA to make proteins. Messenger RNA molecules carry the genetic information from the DNA in the nucleus of the cell to the cytoplasm where the proteins are made. Messenger RNA acts as the messenger to build proteins. "Actually, there are many RNAs (ribonucleic acids) that are not used to make proteins," explained Chen. "About 70 percent of the human genome is used to make noncoding RNAs that don't code for protein sequences but have other uses." Telomerase RNA is one of the noncoding RNAs that assembles along with telomerase proteins to form the enzyme telomerase. Telomerase is crucial for cellular immortality in cancer and stem cells. In this study, Chen's group shows that a fungal telomerase RNA is processed from a protein-coding mRNA, instead of being synthesized independently. "Our finding from this paper is paradigm-shifting. Most RNA molecules are synthesized independently and here we uncovered a dual function mRNA that can be used to produce a protein or to make a noncoding telomerase RNA, which is really unique," said Chen. "We will need to do a lot more research to understand the underlying mechanism of such an unusual RNA biogenesis pathway." Basic research on the metabolism and regulation of mRNA has led to important medical applications. For example, several COVID-19 vaccines use messenger RNA as a means to produce viral spike proteins. In these vaccines, the mRNA molecules are eventually degraded and then absorbed by our bodies. This new approach has advantages over DNA vaccines which run the potential risk of being deleteriously and permanently incorporated into our DNA. The discovery of dual-function mRNA biogenesis in this work might lead to innovative ways of making future mRNA vaccines. In this study Chen's group discovered the unexpected mRNA-derived telomerase RNA in the model fungal organism Ustilago maydis or corn smut. Corn smut, also called Mexican truffle, is edible and adds a delicious umami effect to many dishes, for example tamales and tacos. The study of RNA and telomere biology in corn smut may provide opportunities for finding novel mechanisms for mRNA metabolism and telomerase biogenesis. If this mechanism is found to exist in humans, it may have major implications for vaccine development and aging research.
|
|
Scooped by
Dr. Stefan Gruenwald
September 29, 2022 12:14 PM
|
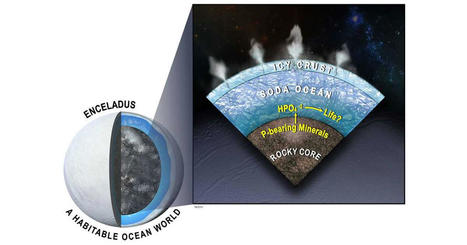
The search for extraterrestrial life just got more interesting as a team of scientists including Southwest Research Institute's Dr. Christopher Glein has discovered new evidence for a key building block for life in the subsurface ocean of Saturn's moon Enceladus. New modeling indicates that Enceladus's ocean should be relatively rich in dissolved phosphorus, an essential ingredient for life. "Enceladus is one of the prime targets in humanity's search for life in our solar system," said Glein, a leading expert in extraterrestrial oceanography. He is a co-author of a paper in the Proceedings of the National Academy of Sciences (PNAS) describing this research. "In the years since NASA's Cassini spacecraft visited the Saturn system, we have been repeatedly blown away by the discoveries made possible by the collected data." The Cassini spacecraft discovered Enceladus's subsurface liquid water and analyzed samples as plumes of ice grains and water vapor erupted into space from cracks in the moon's icy surface. "What we have learned is that the plume contains almost all the basic requirements of life as we know it," Glein said. "While the bioessential element phosphorus has yet to be identified directly, our team discovered evidence for its availability in the ocean beneath the moon's icy crust." One of the most profound discoveries in planetary science over the past 25 years is that worlds with oceans beneath a surface layer of ice are common in our solar system. Such worlds include the icy satellites of the giant planets, such as Europa, Titan and Enceladus, as well as more distant bodies like Pluto. Worlds like Earth with surface oceans must reside within a narrow range of distances from their host stars to maintain the temperatures that support surface liquid water. Interior water ocean worlds, however, can occur over a much wider range of distances, greatly expanding the number of habitable worlds likely to exist across the galaxy. "The quest for extraterrestrial habitability in the solar system has shifted focus, as we now look for the building blocks for life, including organic molecules, ammonia, sulfur-bearing compounds as well as the chemical energy needed to support life," Glein said. "Phosphorus presents an interesting case because previous work suggested that it might be scarce in the ocean of Enceladus, which would dim the prospects for life." Phosphorus in the form of phosphates is vital for all life on Earth. It is essential for the creation of DNA and RNA, energy-carrying molecules, cell membranes, bones and teeth in people and animals, and even the sea's microbiome of plankton. Team members performed thermodynamic and kinetic modeling that simulates the geochemistry of phosphorus based on insights from Cassini about the ocean-seafloor system on Enceladus. In the course of their research, they developed the most detailed geochemical model to date of how seafloor minerals dissolve into Enceladus's ocean and predicted that phosphate minerals would be unusually soluble there. "The underlying geochemistry has an elegant simplicity that makes the presence of dissolved phosphorus inevitable, reaching levels close to or even higher than those in modern Earth seawater," Glein said. "What this means for astrobiology is that we can be more confident than before that the ocean of Enceladus is habitable." According to Glein, the next step is clear: "We need to get back to Enceladus to see if a habitable ocean is actually inhabited."
|
|
Scooped by
Dr. Stefan Gruenwald
August 27, 2022 5:49 PM
|
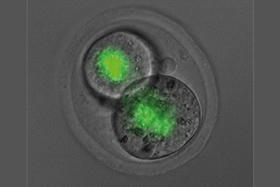
A fundamental discovery about a driver of healthy development in embryos could rewrite our understanding of what can be inherited from our parents and how their life experiences may shape us. The new research suggests that epigenetic information, which sits on top of DNA and is normally reset between generations, is more frequently carried from mother to offspring than previously thought. The new study significantly broadens our understanding of which genes have epigenetic information passed from mother to child and which proteins are important for controlling this unusual process. At a glance - First study to find a protein in the mother’s egg that regulates the epigenetic inheritance of a set of genes critical for the development of normal body structure in mammals.
- While the epigenome can be influenced by the environment, including someone’s diet and exposure to pollutants, these epigenetic changes are very rarely inherited.
- Discovery transforms our understanding of what can be passed down, indicating epigenetic inheritance may occur more frequently than previously thought.
Epigenetics is a rapidly growing field of science that investigates how our genes are switched on and off to allow one set of genetic instructions to create hundreds of different cell types in our body. Epigenetic changes can be influenced by environmental variations such as our diet, but these changes do not alter DNA and are normally not passed from parent to offspring. While a tiny group of ‘imprinted’ genes can carry epigenetic information across generations, until now, very few other genes have been shown to be influenced by the mother’s epigenetic state. The new research reveals that the supply of a specific protein in the mother’s egg can affect the genes that drive skeletal patterning of offspring. Chief investigator Professor Marnie Blewitt said the findings initially left the team surprised. “It took us a while to process because our discovery was unexpected,” Professor Blewitt, Joint Head of the Epigenetics and Development Division at WEHI, said.
|
|
Scooped by
Dr. Stefan Gruenwald
August 11, 2022 10:26 PM
|
Classically, light microscopes limits are in the micrometer range. Advances in single-molecule localization microscopy allow for nanometer resolution. Improving the resolution of optical microscopes was thought to be limited by a theorem put forward in the 19th century. However, scientists have been able to overcome this issue in the 21stcentury. In the 16-17th century Europe, a Dutch tradesman and scientist named Antoine van Leeuwenhoek used microscopes to discover red blood cells and to first see living sperm cells of animals. In 1873, there was an equation published by Ernst Abbe demonstrating how the resolution of a light microscope was limited by, among other things, the wavelength of light. Through the 19th and much of the 20th century this limit, Abbe’s Limit (0.2 um), was thought to be unsurpassable. However, in 2014, the Nobel Prize in Chemistry was awarded for a technique called single-molecule localization microscopy (SMLM) which bypasses Abbe’s diffraction limit and set a new precedent for the field of microscopy—resolution at the nanoscale level. Today, SMLM approaches have the potential to capture natural processes at the nanoscale level such as molecular complexes, protein-protein interactions, and spatial organizations. A recent work by Coelho et al. leverages this technique, improves upon it, and uses it to look closely at an immune cell called a T cell. The two primary super resolution techniques are stochastic optical reconstruction microscopy (STORM) and DNA point accumulation for imaging in nanoscale topography (DNA-PAINT). In SMLM, single fluorophores are temporally separated by using a weak light pulse to activate a fraction of all the fluorophores. These fluorophores glow until bleached at which point the process is repeated for a new set of fluorophores. Next, the blurry images are mathematically processed using probability theory (i.e. where is the original point source of the light?) to render them much sharper into points. Eventually every frame is superimposed to form one final, super resolution image. However, the time required to sequential image all the individual fluorophores require long acquisition times, as typically tens of thousands of frames are needed to map a given protein species in a cell. Drift during camera exposure affects the localization precision and the accuracy of localization. Thus, in practice the resolution is reduced to tens of nanometers making it unfeasible to conduct distance measurements on biological relevant scales. Coelho et al. overcome this issue using an engineering solution coined Feedback SMLM, “…which can capture molecular emissions in a complex and unknown cellular environment with equal probability and high precision.” In other words, their technique can measure distances between two proteins in a wide range of sample formats, including intact cells, to capture heterogeneity as well as rare events with nanometer resolution. Feedback SMLM uses three types of corrections: stage-sample feedback loop, autonomous optical feedback loop and piezoelectric mirror correction. Stage-sample feedback is accomplished by using non-fluorescent fiducials (reference points) outside of the field of view. A camera operating at a speed of 370 frames per second tracks the fiducials to provide information of both x-y and z position. Stage corrections can happen during sample acquisition with stabilization of 0.4 and 1 nm (SD) in the lateral (x-y) and axial (z) directions, respectively, over hours and days. Optical feedback uses a white LED integrated in the microscope body, creating an optical fiducial, which a camera is monitoring with a precision of 0.05 nm. Finally, the piezoelectric mirror tracks the mechanical instabilities and reduces image drift to 0.22 nm. The mirror correction accomplishes something other SMLM techniques can not; that is the stability of fluorescence path does not have to be computationally corrected post-acquisition. Using DNA-PAINT with Feedback SMLM the authors generate an ultrahigh resolution image of F-actin. F-actin is an abundant protein in cells and used as an imaging benchmark because the width of actin is sub diffraction limit (i.e <0.2 um). Incredibly an individual actin fibril can be accurately measured to width of 5 to 9 nm without any post-acquisition processing. Electron microscopy measurements corroborate these results. In contrast other current SMLM imaging approaches, e.g. dSTORM, were only able to obtain an approximate width of 20 nm due to insufficient localization precision. Finally, the authors apply their technique to an unanswered immunological question: how do T cells (derived from the Thymus) signal to become activated? T cells activate through their T cell receptor (TCR) which recognizes peptides presented in major histocompatibility complex (pMHC). T cells are exquistively sensitive and can recognize fewer than 10 agonist pMHC ligands with only a few TCRs. The current model suggests a phosphorylation cascade on associated protein dimers (i.e. CD3 dimers) of the TCR triggers cellular activation. It is widely assumed TCR triggering requires the exclusion of the transmembrane phosphatase CD45 to successfully activate. However, these delicate events have never been visualized before and are currently readout biochemically. Coelho et al. activated Jurkat T cells using pMHC coated on a lipid bilayer glass slide. The authors applied Feedback SMLM and DNA-PAINT to monitor CD45 and phosphorylated CD3ζ (pCD3ζ). What they found was spatially separated nano-clusters of pCD3ζ and CD45 at median distances of 19.6 nm. On resting T cells CD3 and CD45 appeared intermix with mean distances of 12.5 nm. Thus, if spatial separation was the means of TCR triggering, the TCR is exquisitely sensitive to 4- to 7-nm differences between CD3 and CD45. Through careful microscopy image analysis, the author's feedback SMLM has provided the means to answer a question previously unknown due to technological reasons and provides new insights into the field of T cell biology. Reference 1) Coelho et al. Ultraprecise single-molecule localization microscopy enables in situ distance measurements in intact cells. Science Advances. Vol 6, Issue 16. 2020. DOI: 10.1126/sciadv.aay8271
|
|
Scooped by
Dr. Stefan Gruenwald
August 6, 2022 1:46 PM
|
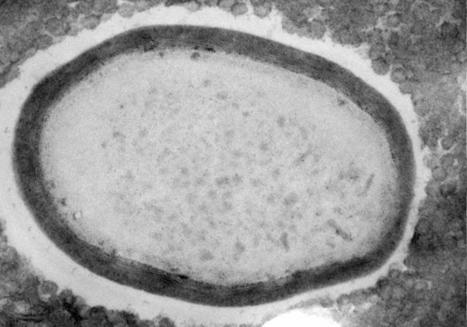
A research team led by André Marques at the Max Planck Institute for Plant Breeding Research in Cologne, Germany, has uncovered the profound effects of an atypical mode of chromosome arrangement on genome organization and evolution. Their findings are published in the journal Cell. In each individual cell in our body, our DNA, the molecule carrying the instructions for development and growth, is packaged together with proteins into structures called chromosomes. Full sets of chromosomes together constitute the genome, the entire genetic information of an organism. In most organisms, including us, chromosomes appear as X-shaped structures when they are captured in their condensed, duplicated states in preparation for cell division. Indeed, these structures may be among the most iconic in all of science. The X shape is due to a constricted region called the centromere that serves to connect sister chromatids, which are the identical copies formed by the DNA replication of a chromosome. Most studied organisms are 'monocentric', meaning that centromeres are restricted to a single region on each chromosome. Several animal and plant organisms, however, show a very different centromere organization: instead of one solitary constriction as in the classic X-shaped chromosomes, chromosomes in these organisms harbor multiple centromeres that are arranged in a line from one end of a sister chromatid to the other. Thus, these chromosomes lack a primary constriction and the X shape, and species with such chromosomes are known as 'holocentric', from the ancient Greek word hólos meaning 'whole'. A new study led by André Marques from the Max Planck Institute for Plant Breeding Research in Cologne, Germany, now reveals the striking effects of this non-classical mode of chromosome organization on genome architecture and evolution. To determine how holocentricity affects the genome, Marques and his team used highly accurate DNA sequencing technology to decode the genomes of three closely related holocentric beak-sedges, grass-like flowering plants found worldwide that are often the first conquerors of new habitats. For reference, the team also decoded the genome of their most closely related monocentric relative. Thus, comparing the holocentric beak-sedges with their monocentric relative allowed the authors to attribute any differences they observed to the effects of holocentricity. Their analysis reveals striking differences in genome organization and chromosome behavior in holocentric organisms. They found that centromere function is distributed across hundreds of small centromere domains in holocentric chromosomes. While in monocentric organisms, genes are largely concentrated distant from centromeres and the regions immediately around them, in holocentric species they are uniformly distributed over the whole length of chromosomes. Further, in monocentric species chromosomes are known to engage in a high degree of intermingling with each other during cell division, a property which appears to play a role in regulating gene expression. Notably, these long-range interactions were sharply diminished in the beak-sedges with holocentromeres. Thus, holocentricity fundamentally affects genome organization as well as how chromosomes behave during cell division. In holocentric organisms, almost any given chromosomal fragment will harbor a centromere and will thus have proper centromere function, which is not true for monocentric species. In this way, holocentromeres have been thought to stabilize chromosomal fragments and fusions and thus promote rapid genome evolution, or the ability of an organism to make prompt, wholesale changes to its DNA. In one of the beak-sedges they analyzed, Marques and his team could show that chromosome fusions facilitated by holocentromeres allowed this species to maintain the same chromosome number even after quadruplication of the entire genome. In another of their analyzed beak-sedges, a species with only two chromosomes, the lowest of any plant, holocentricity was found to be responsible for the dramatic reduction in chromosome number. Thus, holocentric chromosomes may allow the formation of news species through rapid evolution at genome-level.
|
|
Rescooped by
Dr. Stefan Gruenwald
from Virus World
August 1, 2022 1:14 PM
|
Ancient genomes from the herpes virus that commonly causes lip sores – and currently infects some 3.7 billion people globally – have been uncovered and sequenced for the first time by an international team of scientists led by the University of Cambridge. Latest research suggests that the HSV-1 virus strain behind facial herpes as we know it today arose around five thousand years ago, in the wake of vast Bronze Age migrations into Europe from the Steppe grasslands of Eurasia, and associated population booms that drove rates of transmission. Herpes has a history stretching back millions of years, and forms of the virus infect species from bats to coral. Despite its contemporary prevalence among humans, however, scientists say that ancient examples of HSV-1 were surprisingly hard to find. The authors of the study, published in the journal Science Advances, say the Neolithic flourishing of facial herpes detected in the ancient DNA may have coincided with the advent of a new cultural practice imported from the east: romantic and sexual kissing. “The world has watched COVID-19 mutate at a rapid rate over weeks and months. A virus like herpes evolves on a far grander timescale,” said co-senior author Dr Charlotte Houldcroft, from Cambridge’s Department of Genetics. “Facial herpes hides in its host for life and only transmits through oral contact, so mutations occur slowly over centuries and millennia. We need to do deep time investigations to understand how DNA viruses like this evolve,” she said. “Previously, genetic data for herpes only went back to 1925.” The team managed to hunt down herpes in the remains of four individuals stretching over a thousand-year period, and extract viral DNA from the roots of teeth. Herpes often flares up with mouth infections: at least two of the ancient cadavers had gum disease and a third smoked tobacco. The oldest sample came from an adult male excavated in Russia’s Ural Mountain region, dating from the late Iron Age around 1,500 years ago. Two further samples were local to Cambridge, UK. One a female from an early Anglo-Saxon cemetery a few miles south of the city, dating from 6-7th centuries AD. The other a young adult male from the late 14th century, buried in the grounds of medieval Cambridge’s charitable hospital (later to become St. John’s College), who had suffered appalling dental abscesses. The final sample came from a young adult male excavated in Holland: a fervent clay pipe smoker, most likely massacred by a French attack on his village by the banks of the Rhine in 1672. “We screened ancient DNA samples from around 3,000 archaeological finds and got just four herpes hits,” said co-lead author Dr Meriam Guellil, from Tartu University’s Institute of Genomics. “By comparing ancient DNA with herpes samples from the 20th century, we were able to analyse the differences and estimate a mutation rate, and consequently a timeline for virus evolution,” said co-lead author Dr Lucy van Dorp, from the UCL Genetics Institute. Co-senior author Dr Christiana Scheib, Research Fellow at St. John’s College, University of Cambridge, and Head of the Ancient DNA lab at Tartu University, said: “Every primate species has a form of herpes, so we assume it has been with us since our own species left Africa.” “However, something happened around five thousand years ago that allowed one strain of herpes to overtake all others, possibly an increase in transmissions, which could have been linked to kissing.” The researchers point out that the earliest known record of kissing is a Bronze Age manuscript from South Asia, and suggest the custom – far from universal in human cultures – may have travelled westward with migrations into Europe from Eurasia. In fact, centuries later, the Roman Emperor Tiberius tried to ban kissing at official functions to prevent disease spread, a decree that may have been herpes-related. However, for most of human prehistory, HSV-1 transmission would have been “vertical”: the same strain passing from infected mother to newborn child. Two-thirds of the global population under the age of 50 now carry HSV-1, according to the World Health Organization. For most of us, the occasional lip sores that result are embarrassing and uncomfortable, but in combination with other ailments – sepsis or even COVID-19, for example – the virus can be fatal. In 2018, two women died of HSV-1 infection in the UK following Caesarean births. “Only genetic samples that are hundreds or even thousands of years old will allow us to understand how DNA viruses such as herpes and monkeypox, as well as our own immune systems, are adapting in response to each other,” said Houldcroft. The team would like to trace this hardy primordial disease even deeper through time, to investigate its infection of early hominins. “Neanderthal herpes is my next mountain to climb,” added Scheib. Publisjhed in Science Advances (July 27, 2022): https://doi.org/10.1126/sciadv.abo4435
Via Juan Lama
|
|
|
Rescooped by
Dr. Stefan Gruenwald
from Virus World
February 20, 2023 5:25 PM
|
A study employing CRISPR/Cas9 to explore the evolutionary beginnings of some giant viruses finds evidence that their large genomes arose from gene duplications. Most viruses are small and carry minimal genomes. Even one of the largest small viruses, Vaccinia, measures merely one-fiftieth the size of a pollen grain and contains only 270 genes. Giant viruses flout these rules. With sizes that rival small bacteria and genomes that contain thousands of genes, their complexity emulates that of cellular life. How these viruses came to be so large has been the subject of much debate. Now, scientists are finally poised to unravel the mystery of their evolutionary origins, thanks to a suite of CRISPR/Cas9-based tools described in a Nature Communications paper from January. “It was by chance that we encountered the first giant virus,” says Chantal Abergel, a virologist at Aix-Marseille University in France. “It was Mimivirus, and it was actually mistaken for a bacterium.” In the 20 years since that discovery, virologists have prioritized exploring the diversity of giant viruses. Now that they’ve found a fair few, the focus has shifted towards studying their evolution in more detail with molecular biology techniques. Evolutionary biologists have grappled over two possible origins of giant viruses. One possibility is that they were once cellular organisms that shrunk physically and genetically over time. But most virologists now suspect giant viruses grew out of much smaller ones—though the evidence supporting either hypothesis is scant. To begin addressing this origin question, Abergel decided to examine how the essential genes in the Pandoravirus genome are distributed. In cellular organisms, essential genes are scattered throughout the genome—so if giant viruses are essentially reduced cells, one would expect a similar pattern. Alternatively, if the genes are clumped, that could indicate the viruses’ large genomes started out in a more compact form. One way to locate a virus’s essential genes is to knock out genes one at a time to find the ones that are needed for virus production. But to do that with a giant virus, Abergel needed a gene-editing system that worked in members of the group. With the help of Hugo Bisio, a postdoctoral researcher in Abergel’s lab, and colleagues at Aix–Marseille University, Abergel used a CRISPR/Cas9-based gene-editing system to modify the genome of the amoeba Acanthamoeba castellanii and the giant virus Pandoravirus neocaledonia, which infects it. The CRISPR/Cas9 system was designed to delete specific genes and consists of two guide RNAs and a Cas9 scission enzyme. Similar to other CRISPR/Cas9 systems, each guide RNA contains 17 to 20 bases designed to bind to one specific location on the genome of the giant virus or the amoeba, allowing the Cas9 scission enzyme to cut the genome at that site. The amoeba A. castellanii contains 25 copies of each chromosome, making it difficult to design an efficient CRISPR/Cas9 system that could delete each gene copy. To overcome this issue, the researchers modified their CRISPR/Cas9 system to generate a chain reaction. Each time DNA was cut to remove a gene, a DNA segment encoding the Cas9 enzyme and the guide RNAs responsible for the cut would take the place of the missing gene in the genome. This allowed gene deletions to repeat and propagate until all copies were removed. Once they optimized their CRISPR/Cas9 system, the team deleted each gene separately from the Pandoravirus genome and measured the resulting change in virus production, in order to determine how important each gene is to the virus’s lifecycle. They found that essential genes clustered together at one end of the genome and were segregated from nonessential genes at the other end. This level of gene orderliness has not been seen in viruses, according to Bisio. Even bacterial genomes aren’t quite so tidy: While they do group genes with linked functions together into gene clusters known as operons, these tend to be dispersed throughout the genome rather than grouped all together in one spot. Bisio says the cluster of essential genes may echo a smaller “core genome” of an ancient virus. This genome could have become elongated through multiple rounds of gene duplication that were biased in one direction to produce an additional set of spare nonessential genes. This could explain how modern-day giant viruses came to possess thousands of genes. “Our data indicate that complex viruses arose from smaller and simpler ones,” Bisio tells The Scientist in an email—noting that it will take further research to determine whether that’s true of all giant viruses or just Pandoravirus. Other studies found that some genes in giant viruses were usurped from their amoeba hosts, suggesting gene exchange is another way giant viruses increased in size. The team then set their sights on one of the many evolutionary mysteries of Pandoravirus: its lack of a capsid. Small viruses package their genomes into capsids made of viral proteins. While some giant viruses, such as Mimivirus, continue this tradition, others, including Pandoravirus, do not. If giant viruses did indeed evolve from smaller ones, there could be traces of capsid proteins hiding in their genomes. So, the researchers set out to study the function of potential capsid protein remnants in a close cousin of Pandoravirus, the smaller Mollivirus, which can also infect A. castellanii. Researchers have suspected that a Mollivirus protein called ml_347 evolved from a capsid gene based on its gene’s sequence and predicted 3D shape. So, the team investigated its function by deleting the gene using their CRISPR/Cas9 system. They found that the gene is important for Mollivirus assembly, which the authors say is intriguing given its possible capsid ancestry. It’s possible that, as capsids were lost in giant virus evolution, obsolete capsid genes were adapted for new assembly functions. Frederik Schulz, an evolutionary biologist with the DOE Joint Genome Institute in California who wasn’t involved with the study but who has worked with Chantal Abergel in the past, tells The Scientist that the findings align with recent discoveries. “There was a debate for a long time [about] how giant virus[es] evolved,” he says. “The working hypothesis in the previous years was that they evolved from smaller viruses, and that’s exactly what Chantal and her team could show and confirm using their CRISPR/Cas9 gene-editing approach.” Schulz notes that it will be exciting to see the CRISPR/Cas9 technology introduced into other host species, such as algae, which would allow researchers to expand research into a greater variety of giant viruses. He also points out that the system only works for viruses that replicate in a host cell’s nucleus, while most giant viruses replicate in cytoplasmic structures called viral factories, which the Cas9 enzyme and guide RNAs can’t penetrate. Still, Bisio says there’s much left to discover in Pandoravirus. “[This CRISPR/Cas9 technology is] a goldmine to find new functions,” he says—one that he and his colleagues are eager to employ to tease apart what all the virus’s genes do. Cited research published in Nat. Comm. (Jan. 26, 2023): https://doi.org/10.1038/s41467-023-36145-4
Via Juan Lama
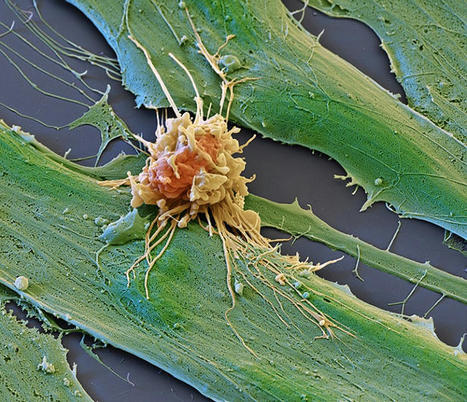
Precision-controlled CAR-T-cell immunotherapies could be used to tackle a range of tumor types. Elaborately engineered immune cells can not only recognize cancer cells, but also evade defenses that tumors use to fend off attacks, researchers have found. Two studies recently published in Science1,2 build on the success of chimeric antigen receptor (CAR)-T cancer therapies, which use genetically altered T cells to seek out tumors and mark them for destruction. These treatments have the potential to lead to long-lasting remission, but are not successful for everyone, and have so far been effective against only a small number of cancers. To bolster the power of CAR-T therapies, researchers have further engineered the cells to contain switches that allow control over when and where the cells are active. The hacked cells produce a protein that stimulates T cells, to counteract immunosuppressive signals that are often released by tumors. Both studies are a tour de force in T-cell engineering and highlight the direction that researchers want to push CAR-T-cell therapy, says systems immunologist Grégoire Altan-Bonnet at the US National Cancer Institute. “We know a lot of the parts, now it’s being able to put them together and explore,” he says. “If we engineer the system well, we can really put the tumors into checkmate.” Engineered immune cells T cells typically patrol the body, looking for foreign proteins displayed on the surface of cells. Such cells could be infected with a virus, for example, or they could be tumor cells that are producing abnormal, cancer-associated proteins. A class of T cells called killer T cells can then destroy the abnormal cells. CAR-T therapies involve genetically engineering T cells from a person with cancer to produce CARs, which are proteins that recognize the proteins displayed by tumor cells. The approach has been approved to treat some leukemias, lymphomas and myelomas. But researchers have been pursuing ways to make the treatments safer and more effective, and to expand their use to other diseases. Highly mutated cancers respond better to immune therapy In one of the new studies, Ahmad Khalil, a synthetic biologist at Boston University in Massachusetts, and his colleagues wired a complex system of 11 DNA sequences into CAR T cells. The resulting genetic circuits can be switched on and off using already-approved drugs, which allows researchers to control when and where the hacked T cells are active, as well as their production of a protein called IL-2, which stimulates immune responses. The other group of researchers, led by synthetic biologist Wendell Lim at the University of California, San Francisco, programmed CAR T cells to produce IL-2 only when the engineered T cell encounters a cancer cell. The team found that this IL-2 production was most efficient at fighting tumors in mice with pancreatic cancer when it was activated through a pathway that was separate from the one used to recognize the cancer cell — a detail that could help in shaping future therapies, says Andrea Schietinger, a tumor immunologist at Memorial Sloan Kettering Cancer Center in New York City.
Via BigField GEG Tech, Gilbert C FAURE
|
|
Scooped by
Dr. Stefan Gruenwald
December 1, 2022 1:47 PM
|
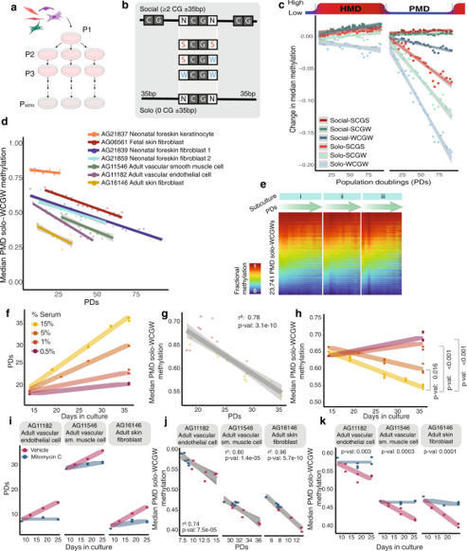
DNA methylation undergoes dramatic age-related changes, first described more than four decades ago. Loss of DNA methylation within partially methylated domains (PMDs), late-replicating regions of the genome attached to the nuclear lamina, advances with age in normal tissues, and is further exacerbated in cancer. Researchers now show experimental evidence that this DNA hypomethylation is directly driven by proliferation-associated DNA replication. Within PMDs, loss of DNA methylation at low-density CpGs in A:T-rich immediate context (PMD solo-WCGWs) tracks cumulative population doublings in primary cell culture. Cell cycle deceleration results in a proportional decrease in the rate of DNA hypomethylation. Blocking DNA replication via Mitomycin C treatment halts methylation loss. Loss of methylation continues unabated after TERT immortalization until finally reaching a severely hypomethylated equilibrium. Ambient oxygen culture conditions increases the rate of methylation loss compared to low-oxygen conditions, suggesting that some methylation loss may occur during unscheduled, oxidative damage repair-associated DNA synthesis. Also, they were able to validate a model which estimates the relative cumulative replicative histories of human cells, which they call “RepliTali” (Replication Times Accumulated in Lifetime). DNA methylation loss has been observed in aging tissues and cancers for decades. Researchers from Van Andel Institute have now provided experimental evidence that this process is directly driven by cell division, something that hasn't been directly shown before.
|
|
Rescooped by
Dr. Stefan Gruenwald
from Virus World
November 24, 2022 3:52 PM
|
Phages probably picked up DNA-cutting systems from their microbial hosts, and may use them to fight other viruses. A systematic sweep of viral genomes has revealed a trove of potential CRISPR-based genome-editing tools. CRISPR–Cas systems are common in the microbial world of bacteria and archaea, where they often help cells to fend off viruses. But an analysis1 published on 23 November 2022 in Cell finds CRISPR–Cas systems in 0.4% of publicly available genome sequences from viruses that can infect these microbes. Researchers think that the viruses use CRISPR–Cas to compete with one another — and potentially also to manipulate gene activity in their host to their advantage. Some of these viral systems were capable of editing plant and mammalian genomes, and possess features — such as a compact structure and efficient editing — that could make them useful in the laboratory. “This is a significant step forward in the discovery of the enormous diversity of CRISPR–Cas systems,” says computational biologist Kira Makarova at the US National Center for Biotechnology Information in Bethesda, Maryland. “There is a lot of novelty discovered here.” DNA-cutting defenses Although best known as a tool used to alter genomes in the laboratory, CRISPR–Cas can function in nature as a rudimentary immune system. About 40% of sampled bacteria and 85% of sampled archaea have CRISPR–Cas systems. Often, these microbes can capture pieces of an invading virus’s genome, and store the sequences in a region of their own genome, called a CRISPR array. CRISPR arrays then serve as templates to generate RNAs that direct CRISPR-associated (Cas) enzymes to cut the corresponding DNA. This can allow microbes carrying the array to slice up the viral genome and potentially stop viral infections. Viruses sometimes pick up snippets of their hosts’ genomes, and researchers had previously found isolated examples of CRISPR–Cas in viral genomes. If those stolen bits of DNA give the virus a competitive advantage, they could be retained and gradually modified to better serve the viral lifestyle. For example, a virus that infects the bacterium Vibrio cholera uses CRISPR–Cas to slice up and disable DNA in the bacterium that encodes antiviral defences2. Molecular biologist Jennifer Doudna and microbiologist Jillian Banfield at the University of California, Berkeley, and their colleagues decided to do a more comprehensive search for CRISPR–Cas systems in viruses that infect bacteria and archaea, known as phages. To their surprise, they found about 6,000 of them, including representatives of every known type of CRISPR–Cas system. “Evidence would suggest that these are systems that are useful to phages,” says Doudna. The team found a wide range of variations on the usual CRISPR–Cas structure, with some systems missing components and others unusually compact. “Even if phage-encoded CRISPR–Cas systems are rare, they are highly diverse and widely distributed,” says Anne Chevallereau, who studies phage ecology and evolution at the French National Centre for Scientific Research in Paris. “Nature is full of surprises.” Small, but efficient Viral genomes tend to be compact, and some of the viral Cas enzymes were remarkably small. This could offer a particular advantage for genome-editing applications, because smaller enzymes are easier to shuttle into cells. Doudna and her colleagues focused on a particular cluster of small Cas enzymes called Casλ, and found that some of them could be used to edit the genomes of lab-grown cells from thale cress (Arabidopsis thaliana), wheat, as well as human kidney cells. The results suggest that viral Cas enzymes could join a growing collection of gene-editing tools discovered in microbes. Although researchers have uncovered other small Cas enzymes in nature, many of those have so far been relatively inefficient for genome-editing applications, says Doudna. By contrast, some of the viral Casλ enzymes combine both small size and high efficiency. In the meantime, researchers will continue to search microbes for potential improvements to known CRISPR–Cas systems. Makarova anticipates that scientists will also be looking for CRISPR–Cas systems that have been picked up by plasmids — bits of DNA that can be transferred from microbe to microbe. “Each year we have thousands of new genomes becoming available, and some of them are from very distinct environments,” she says. “So it’s really going to be interesting.” Published in Nature (Nov. 23, 2022): https://doi.org/10.1038/d41586-022-03837-8
Via Juan Lama
|
|
Scooped by
Dr. Stefan Gruenwald
November 1, 2022 6:58 PM
|
Microbial molecules from soil, seawater and human bodies are among the planet’s least understood proteins. The ESM Metagenomic Atlas database contains structure predictions for 617 million proteins. When London-based Deep Mind unveiled predicted structures for some 220 million proteins this year, it covered nearly every protein from known organisms in DNA databases. Now, another tech giant is filling in the dark matter of our protein universe.
Researchers at Meta (formerly Facebook) have used artificial intelligence (AI) to predict the structures of some 600 million proteins from bacteria, viruses and other microbes that haven’t been characterized. ‘It will change everything’: DeepMind’s AI makes gigantic leap in solving protein structures
“These are the structures we know the least about. These are incredibly mysterious proteins. I think they offer the potential for great insight into biology,” says Alexander Rives, the research lead for Meta AI’s protein team.
The team generated the predictions — described in a November 2022 preprint — using a ‘large language model’, a type of AI that are the basis for tools that can predict text from just a few letters or words.
Normally language models are trained on large volumes of text. To apply them to proteins, Rives and his colleagues fed them sequences to known proteins, which can be expressed by a chains of 20 different amino acids, each represented by a letter. The network then learned to ‘autocomplete’ proteins with a proportion of amino acids obscured.
Protein ‘autocomplete’
This training imbued the network with an intuitive understanding of protein sequences, which hold information about their shapes, says Rives. A second step — inspired by DeepMind’s pioneering protein structure AI AlphaFold — combines such insights with information about the relationships between known protein structures and sequences, to generate predicted structures from protein sequences.
Meta’s network, called ESMFold, isn’t quite as accurate as AlphaFold, Rives’ team reported earlier this summer2, but it is about 60 times faster at predicting structures, he says. “What this means is that we can scale structure prediction to much larger databases.”
As a test case, they decided to wield their model on a database of bulk-sequenced ‘metagenomic’ DNA from environmental sources including soil, seawater, the human gut, skin and other microbial habitats. The vast majority of the DNA entries — which encode potential proteins — come from organisms that have never been cultured and are unknown to science.
In total, the Meta team predicted the structures of more than 617 million proteins. The effort took just 2 weeks (AlphaFold can take minutes to generate a single prediction). The predictions are freely available for anyone to use, as is the code underlying the model, says Rives. Of these 617 million predictions, the model deemed more than one-third to be high quality, such that researchers can have confidence that the overall protein shape is correct and, in some cases, can discern finer atomic-level details. Millions of these structures are entirely novel, and unlike anything in databases of protein structures determined experimentally or in the AlphaFold database of predictions from known organisms.
A good chunk of the AlphaFold database is made of structures that are nearly identical to each other, and ‘metagenomic’ databases “should cover a large part of the previously unseen protein universe”, says Martin Steinegger, a computational biologist at Seoul National University. “There’s a big opportunity now to unravel more of the darkness.”
Sergey Ovchinnikov, an evolutionary biologist at Harvard University in Cambridge, Massachusetts, wonders about the hundreds of millions of predictions that ESMFold made with low-confidence. Some might lack a defined structure, at least in isolation, whereas others might be non-coding DNA mistaken as a protein-coding material. “It seems there is still more than half of protein space we know nothing about,” he says.
|
|
Scooped by
Dr. Stefan Gruenwald
October 18, 2022 6:56 PM
|
A new small blue snailfish is changing our understanding of the world's deepest fishes. In 2018, an international team of scientists studied the Atacama Trench, an expansive trench that runs along the west coast of South America as a deep underwater valley that mirrors the Andes Mountains. The team, including Newcastle University scientists, deployed free-falling landers to sample the sparse deep-sea creatures around cameras and traps with bait. Two lander systems from Newcastle University recorded three types of hadal snailfish and one of them was not like the others. The small blue fish, seen from about 6,000 to 7,600 m deep, doesn't look like other hadal snailfish. With large eyes and striking color, it resembles other species of snailfishes that are found living in much shallower waters. The team used a 3D x-ray technique called micro-computed tomography (micro-CT) and DNA barcoding to see where the new species fit within the snailfish family. To the team's surprise, the new species appears to be a separate colonizer of the Atacama Trench. The new species belongs is a member of the genus Paraliparis. Species in this genus are particularly abundant in the Southern Ocean of the Antarctic and are rarely found deeper than 2,000 m. Significantly, this is the first time this genus has been found living in the hadal zone. The team named the new species Paraliparis selti, meaning blue in the Kunza language of the indigenous peoples of the Atacama Desert. The description is published in the journal Marine Biodiversity. Study lead author, Dr Thom Linley, a visiting researcher at Newcastle University said: "I find this family of fishes absolutely fascinating. They are not at all what we expect from a deep-sea fish and I love to show people that the world's deepest fishes are actually pretty cute. "For me to get a camera down to where these animals live, it's made of inches thick stainless steel and sapphire glass. It then films these delicate and beautiful animals perfectly adapted to this extreme environment. With engineering-built force we can only clumsily visit these animals for a short time. We have been wondering for some time just what makes this type of fish so good at living deep. Maybe it was a series of lucky accidents, a chance fluke, that happened in one lineage. Finding this new species tells us that it's bigger than that. Lightning struck twice and there is something special about this Family. Paraliparis selti provides a fantastic opportunity to explore what allows fish to live so deep. If we only had a single lineage to study, we could never be sure which traits were just part of that lineage and which are the deep-sea secret sauce."
|
|
Scooped by
Dr. Stefan Gruenwald
October 4, 2022 4:37 PM
|
New research shows that people with extreme look-alike faces share common genotypes, but differ in their DNA methylation and microbiome landscapes. “For decades, the existence of individuals who resemble each other without having any family ties has been described as a proven fact, but only in anecdotal terms and without any scientific justification,” said senior author Dr. Manel Esteller, a researcher at the Josep Carreras Leukaemia Research Institute. “The widespread use of the Internet and social networks for image-sharing has meant that we are now able to identify and study such people.” In their study, Dr. Esteller and his colleagues set out to characterize a set of look-alike humans on a molecular level. To do so, they recruited human doubles from the photographic work of François Brunelle, a Canadian artist who has been obtaining worldwide pictures of look-alikes since 1999. They obtained headshot pictures of 32 look-alike pairs. They determined an objective measure of likeness for the pairs using three different facial recognition algorithms. In addition, the participants completed a comprehensive biometric and lifestyle questionnaire and provided saliva DNA for multiomics analysis. “This unique set of samples has allowed us to study how genomics, epigenomics, and microbiomics can contribute to human resemblance,” Dr. Esteller said. Overall, the results revealed that these individuals share similar genotypes, but differ in their DNA methylation and microbiome landscapes. Half of the look-alike pairs were clustered together by all three algorithms. Genetic analysis revealed that 9 of these 16 pairs clustered together, based on 19,277 common single-nucleotide polymorphisms. Moreover, physical traits such as weight and height, as well as behavioral traits such as smoking and education, were correlated in look-alike pairs. Taken together, the results suggest that shared genetic variation not only relates to similar physical appearance, but may also influence common habits and behavior. “Our findings provide a molecular basis for future applications in fields such as biomedicine, evolution, and forensics,” Dr. Esteller said. “It would be very interesting to follow up the potential application in forensics, using the genome of unknown people to prepare bioinformatic strategies to reconstruct the face from DNA. And in medicine, we may be able to deduce the genome of a person from facial analysis and hence use this as a pre-screening tool to detect the presence of genetic mutations associated with disease and apply preventive strategies at an early stage. Some previous studies have compared the genome of individuals with face traits, but in the general population,” he added.
|
|
Rescooped by
Dr. Stefan Gruenwald
from Virus World
October 3, 2022 11:51 AM
|
Reports of myocarditis, penile necrosis, and atypical/persistent rash requiring amputation. Be alert for potentially severe manifestations of monkeypox in patients who are immunocompromised or co-infected with HIV, the CDC told healthcare workers on Thursday. "People who are immunocompromised due to HIV or other conditions are at higher risk for severe manifestations of monkeypox than people who are immunocompetent," the agency said in a health advisory to its Health Alert Network, adding that "the HIV status of all sexually active adults and adolescents with suspected or confirmed monkeypox should be determined." Of the patients with severe manifestations "for whom CDC has been consulted," the majority had HIV with CD4 counts below 200 cells/mL, indicating "substantial immunosuppression," the agency noted. These severe manifestations have included cases involving atypical or persistent rash requiring amputation of an extremity; secondary bacterial or fungal infections; lesions in sensitive areas resulting in severe pain or urethral or bowel strictures; and co-morbidities involving organ systems, including myocarditis, transverse myelitis, bowel lesions, penile necrosis, and others. The news arrives as the U.S. records its third known monkeypox-associated death in Ohio. To date, over 25,000 monkeypox cases have been reported in the U.S., 38% of which involved patients co-infected with HIV. Researchers have highlighted monkeypox severity and outcomes in people with HIV before. A recent study published in The Lancet about a 2017-2018 monkeypox outbreak in Nigeria reported seven deaths in a population of 122 patients, with four of the seven co-infected with HIV. More recently, papers on the current outbreak have reported conflicting results about the association between monkeypox severity and HIV co-infection. CDC data involving more than 1,300 monkeypox patients showed a higher rate of hospitalizations for those with HIV (8% vs 3% for those without HIV), with higher rates for those whose HIV was not virally suppressed. Conversely, German researchers reported that the clinical characteristics in HIV-infected versus non-infected monkeypox patients were largely similar. The new CDC advisory recommends that monkeypox treatment include optimization of immune function for people with immunocompromising conditions, such as limiting the use of immunosuppressive medications when "not otherwise clinically indicated," and offering antiretroviral therapy for those living with HIV. Severe immunocompromising conditions can including those with autoimmune disease where immunodeficiency is a clinical component; leukemia or lymphoma, transplant recipients; and those treated with high-dose steroids, alkylating agents, antimetabolites, radiation, or tumor necrosis factor (TNF) inhibitors. On a case-by-case basis, medications such as oral and intravenous tecovirimat (Tpoxx), cidofovir or brincidofovir, and vaccinia immune globulin intravenous should be considered, "although there are no data on effectiveness in treating human monkeypox with these medical countermeasures," according to the CDC. Clinicians should gather repeat lesion swabs in patients with persistent monkeypox DNA, and continue tecovirimat beyond 14 days (but not more than 90 days), "until there is clinical improvement," the advisory stated. Modifications to the dose, frequency, and duration of tecovirimat may be necessary, depending on the patient's clinical condition, disease progression, or therapeutic response. The advisory encouraged clinicians to consult with the CDC Monkeypox Response Clinical Escalations team when appropriate. The following severe manifestations seen in monkeypox patients were reported to the CDC, although the advisory did not include information about HIV status: - Atypical or persistent rash with coalescing or necrotic lesions, or both, some of which have required extensive surgical debridement or amputation of an affected extremity
- Lesions on a significant proportion of the total body surface area, which may be associated with edema and secondary bacterial or fungal infections among other complications
- Lesions in sensitive areas (including mucosal surfaces such as the oropharynx, urethra, rectum, and vagina) resulting in severe pain that interferes with activities of daily living
- Bowel lesions that are exudative or cause significant tissue edema, leading to obstruction
- Severe lymphadenopathy that can be necrotizing or obstructing (such as in airways)
- Lesions leading to stricture and scar formation resulting in significant morbidity such as urethral and bowel strictures, phimosis, and facial scarring
CDC Health Advisory published September 29, 2022
Via Juan Lama
|
|
Rescooped by
Dr. Stefan Gruenwald
from Virus World
August 27, 2022 6:10 PM
|
Low cortisol levels and herpes-virus reactivation are associated with prolonged COVID-19 symptoms, preliminary research suggests. Researchers looking for biological drivers and markers of long COVID have linked the syndrome to herpes viruses, as well as to reduced levels of a stress hormone1. The exploratory study drew both praise and criticism from scientists contacted by Nature. “A major weakness of this paper is the very small sample size,” says infectious-disease specialist Michael Sneller at the US National Institute of Allergy and Infectious Diseases (NIAID) in Bethesda, Maryland. “I would take with a huge grain of salt all these findings unless they are confirmed in larger studies.” But Danny Altmann, an immunologist at Imperial College London who was not involved in the study, says such reports are exactly what the long-COVID research community needs. “We’re really desperate to work out which drugs to test” for long COVID, he says. “The more studies we have like this, the more it enables us to race off and plan appropriate randomized, controlled trials.” The study, posted on the preprint server medRxiv, has not yet been peer reviewed. The disease that lingers Long COVID consists of a constellation of sometimes debilitating symptoms that last for months or years after a SARS-CoV-2 infection. It is common — affecting anywhere from 5% to 50% of people who contract COVID-19 — but researchers have few clues to its causes. The latest study, led by immunobiologist Akiko Iwasaki at Yale School of Medicine in New Haven, Connecticut, involved performing tests that generated thousands of data points for each of 215 participants in an attempt to reveal key immunological features that distinguish people with long COVID from healthy individuals. The study included 99 people with long COVID — those who reported persistent symptoms after SARS-CoV-2 infection — and 116 healthy participants who had either never had COVID-19 or had recovered after SARS-CoV-2 infection. Most strikingly, the study found that in the long-COVID group, levels of cortisol, a stress hormone that has a role in regulating inflammation, blood sugar levels and sleep cycles, were about 50% lower than in healthy participants. The authors also found hints that in people with long COVID, Epstein–Barr virus, which can cause mononucleosis, and varicella-zoster virus, which causes chickenpox and shingles, might recently have been ‘reactivated’. Both of these viruses are in the herpes family, persist indefinitely in the body after infection and can start to multiply again after a period of quiescence. People in the long-COVID group also had unusual levels of certain types of blood and immune cell, as well as dysfunctional immune cells, suggesting that their immune systems are working overtime. Iwasaki cautions that the results do not suggest that low cortisol levels or herpes viruses cause long COVID, but that their association with the syndrome warrants further study. The findings build on previous reports associating low cortisol levels and Epstein–Barr virus with long COVID, including a Cell paper2 published in March. With only 215 participants, the Iwasaki’s study is relatively small, and only a subset of the participants were included in some parts of the work. For example, in the analysis of cortisol levels, the researchers included all 99 people with long COVID, but only 40 of the 116 healthy people. “The small number of controls compared to the long-COVID group is problematic” because it makes the study less statistically robust, says Sneller, who is leading a separate study on the drivers of long COVID. Iwasaki says that samples were collected from the remaining healthy volunteers later and are being sent for analysis this week. Another limitation of the study is that Iwasaki’s team did not analyse participants’ samples for the presence of the Epstein–Barr and varicella-zoster viruses, instead measuring levels of antibodies against the viruses. Members of the long-COVID group had higher levels of these antibodies than did the healthy people, which suggests a more recent reactivation of the virus, Iwasaki says. However, the only way to determine definitively whether there is current reactivation is to test for viral DNA in the blood. Iwasaki’s team is now running such tests. Her group also plans to test participants’ cortisol levels throughout the day, because the stress hormone’s levels tend to fluctuate, and to examine how immune features vary at different time points after infection. “This is an exploratory, hypothesis-generating study, and clearly not the conclusion to what long COVID is, but it does begin to open doors,” says Iwasaki. Published in Nature (August 25, 2022): https://doi.org/10.1038/d41586-022-02296-5
Via Juan Lama
|
|
Rescooped by
Dr. Stefan Gruenwald
from Virus World
August 14, 2022 9:06 PM
|
Human monkeypox virus is spreading in Europe and the USA among individuals who have not travelled to endemic areas. On July 23, 2022, monkeypox was declared a Public Health Emergency of International Concern by WHO Director-General Tedros Adhanom Ghebreyesus. Human-to-human transmission of monkeypox virus usually occurs through close contact with the lesions, body fluids, and respiratory droplets of infected people or animals. The possibility of sexual transmission is being investigated, as the current outbreak appears to be concentrated in men who have sex with men and has been associated with unexpected anal and genital lesions. Whether domesticated cats and dogs could be a vector for monkeypox virus is unknown. Here we describe the first case of a dog with confirmed monkeypox virus infection that might have been acquired through human transmission. Two men who have sex with men attended Pitié-Salpêtrière Hospital, Paris, France, on June 10, 2022 (appendix). One man (referred to as patient 1 going forward) is Latino, aged 44 years, and lives with HIV with undetectable viral loads on antiretrovirals; the second man (patient 2) is White, aged 27 years, and HIV-negative. The men are non-exclusive partners living in the same household. They each signed a consent form for the use of their clinical and biological data, and for the publication of anonymised photographs. The men had presented with anal ulceration 6 days after sex with other partners. In patient 1, anal ulceration was followed by a vesiculopustular rash on the face, ears, and legs; in patient 2, on the legs and back. In both cases, rash was associated with asthenia, headaches, and fever 4 days later. 12 days after symptom onset, their male Italian greyhound, aged 4 years and with no previous medical disorders, presented with mucocutaneous lesions, including abdomen pustules and a thin anal ulceration (figure C, D; appendix). The dog tested positive for monkeypox virus by use of a PCR protocol adapted from Li and colleagues that involved scraping skin lesions and swabbing the anus and oral cavity. Monkeypox virus DNA sequences from the dog and patient 1 were compared by next-generation sequencing (MinION; Oxford Nanopore Technologies, Oxford, UK). Both samples contained virus of the hMPXV-1 clade, lineage B.1, which has been spreading in non-endemic countries since April, 2022, and, as of Aug 4, 2022, has infected more than 1700 people in France, mostly concentrated in Paris, where the dog first developed symptoms. Moreover, the virus that infected patient 1 and the virus that infected the dog showed 100% sequence homology on the 19·5 kilobase pairs sequenced. The men reported co-sleeping with their dog. They had been careful to prevent their dog from contact with other pets or humans from the onset of their own symptoms (ie, 13 days before the dog started to present cutaneous manifestations). In endemic countries, only wild animals (rodents and primates) have been found to carry monkeypox virus. However, transmission of monkeypox virus in prairie dogs has been described in the USA and in captive primates in Europe that were in contact with imported infected animals. Infection among domesticated animals, such as dogs and cats, has never been reported. Published in The Lancet (August 10, 2022):
Via Juan Lama
|
|
Scooped by
Dr. Stefan Gruenwald
August 10, 2022 3:23 PM
|
Epigenomic profiling has enabled large-scale identification of regulatory elements, yet we still lack a systematic mapping from any sequence or variant to regulatory activities. Researchers now address this challenge with Sei, a framework for integrating human genetics data with sequence information to discover the regulatory basis of traits and diseases. Sei learns a vocabulary of regulatory activities, called sequence classes, using a deep learning model that predicts 21,907 chromatin profiles across >1,300 cell lines and tissues. Sequence classes provide a global classification and quantification of sequence and variant effects based on diverse regulatory activities, such as cell type-specific enhancer functions. These predictions are supported by tissue-specific expression, expression quantitative trait loci and evolutionary constraint data. Furthermore, sequence classes enable characterization of the tissue-specific, regulatory architecture of complex traits and generate mechanistic hypotheses for individual regulatory pathogenic mutations. Sei is provided as a resource to elucidate the regulatory basis of human health and disease. Sei is a new framework for integrating human genetics data with a sequence-based mapping of predicted regulatory activities to elucidate mechanisms contributing to complex traits and diseases. Deciphering how regulatory functions are encoded in genomic sequences is a major challenge in understanding how genome variation links to phenotypic traits. Cell type-specific regulatory activities encoded in elements such as promoters, enhancers and boundary elements are critical to defining the complex expression programs essential for multicellular organisms. Most disease-associated variants from genome-wide association studies (GWAS) are located in noncoding regions1, yet without knowing how changes in sequence affect regulatory activities, we cannot predict the impact of these variants and uncover the regulatory mechanisms contributing to complex diseases and traits. Substantial progress has been made in the experimental profiling and integrative analysis of epigenomic marks, such as histone marks and DNA accessibility, across a wide range of tissues and cell types2,3,4. At the same time, deep learning sequence modeling techniques have been successfully applied to learn sequence features predictive of transcription factor (TF) binding and histone modifications5,6,7,8,9,10,11. These models are powerful tools for inferring the impact of sequence variation at the chromatin level—for example, whether a variant increases or decreases C/EBPβ binding. However, we continue to lack an integrative view of sequence regulatory activities, including all major aspects of cis-regulatory functions, such as tissue-specific or broad enhancer and promoter activities. This limits our ability to interpret the integrated effects of all chromatin-level perturbations caused by genomic variants and determine their impact on human health and diseases. The researchers address this challenge by creating a global map for sequence regulatory activity based on a new deep learning-based framework called Sei. This framework introduces a sequence model that predicts 21,907 publicly available chromatin profiles—the broadest set to date—and uses the model to quantitatively characterize regulatory activities for any sequence with a vocabulary we call sequence classes. Sequence classes cover diverse types of regulatory activities, such as promoter or cell type-specific enhancer activity, across the whole genome by integrating sequence-based predictions from histone marks, TFs and chromatin accessibility across a wide range of cell types. Importantly, sequence classes can be used to both classify and quantify the regulatory activities of any sequence based on predictions made by the deep learning sequence model, thereby allowing any mutation to be quantified by its impact (for example, increase, decrease or no change) on cell type-specific regulatory activities. Thus, Sei enables an interpretable and systematic integration of sequence-based regulatory activity predictions with human genetics data to elucidate the regulatory basis of complex traits and diseases. We applied our framework to characterize disease- and trait-associated regulatory disruptions in GWAS data based on a nonoverlapping partitioning of heritability by regulatory activities. Moreover, we applied variant effect prediction at the sequence class-level to interpret cell type-specific regulatory mechanisms for individual disease mutations and differentiate between gain-of-function (GoF) and loss-of-function (LoF) regulatory mutations. The Sei framework in its current form is provided as a resource for systematically classifying and scoring any sequence and variant with sequence classes, additionally providing the Sei model predictions for the 21,907 chromatin profiles underlying the sequence classes. The framework can be run using the code available at https://github.com/FunctionLab/sei-framework; a user-friendly web server is available at hb.flatironinstitute.org/sei.
|
|
Scooped by
Dr. Stefan Gruenwald
August 1, 2022 1:51 PM
|
Constructing a tiny robot from DNA and using it to study cell processes invisible to the naked eye. You would be forgiven for thinking it is science fiction, but it is in fact the subject of serious research by scientists from Inserm, CNRS and Université de Montpellier at the Structural Biology Center in Montpellier. This highly innovative "nano-robot" should enable closer study of the mechanical forces applied at microscopic levels, which are crucial for many biological and pathological processes. It is described in a new study published in Nature Communications. All cells are subject to mechanical forces exerted on a microscopic scale, triggering biological signals essential to many cell processes involved in the normal functioning of our body or in the development of diseases. For example, the feeling of touch is partly conditional on the application of mechanical forces on specific cell receptors (the discovery of which was this year rewarded by the Nobel Prize in Physiology or Medicine). In addition to touch, these receptors that are sensitive to mechanical forces (known as mechanoreceptors) enable the regulation of other key biological processes such as blood vessel constriction, pain perception, breathing or even the detection of sound waves in the ear, etc. The dysfunction of this cellular mechanosensitivity is involved in many diseases -- for example, cancer: cancer cells migrate within the body by sounding and constantly adapting to the mechanical properties of their microenvironment. Such adaptation is only possible because specific forces are detected by mechanoreceptors that transmit the information to the cell cytoskeleton. At present, our knowledge of these molecular mechanisms involved in cell mechanosensitivity is still very limited. Several technologies are already available to apply controlled forces and study these mechanisms, but they have a number of limitations. In particular, they are very costly and do not allow us to study several cell receptors at a time, which makes their use very time-consuming if we want to collect a lot of data. DNA origami structures In order to propose an alternative, the research team led by Inserm researcher Gaëtan Bellot at the Structural Biology Center (Inserm/CNRS/Université de Montpellier) decided to use the DNA origami method. This enables the self-assembly of 3D nanostructures in a pre-defined form using the DNA molecule as construction material. Over the last ten years, the technique has allowed major advances in the field of nanotechnology. This enabled the researchers to design a "nano-robot" composed of three DNA origami structures. Of nanometric size, it is therefore compatible with the size of a human cell. It makes it possible for the first time to apply and control a force with a resolution of 1 piconewton, namely one trillionth of a Newton -- with 1 Newton corresponding to the force of a finger clicking on a pen. This is the first time that a human-made, self-assembled DNA-based object can apply force with this accuracy. The team began by coupling the robot with a molecule that recognizes a mechanoreceptor. This made it possible to direct the robot to some of our cells and specifically apply forces to targeted mechanoreceptors localized on the surface of the cells in order to activate them. Such a tool is very valuable for basic research, as it could be used to better understand the molecular mechanisms involved in cell mechano-sensitivity and discover new cell receptors sensitive to mechanical forces. Thanks to the robot, the scientists will also be able to study more precisely at what moment, when applying force, key signaling pathways for many biological and pathological processes are activated at cell level.
|