Your new post is loading...

|
Scooped by
Dr. Stefan Gruenwald
February 19, 2024 3:12 AM
|
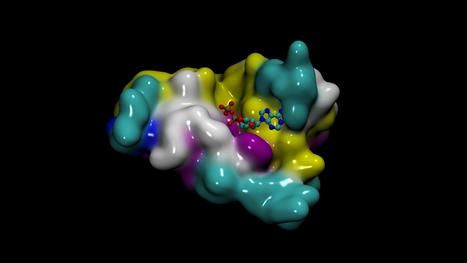
Using a virus-like delivery particle made from DNA, researchers from MIT and the Ragon Institute of MGH, MIT, and Harvard have created a vaccine that can induce a strong antibody response against SARS-CoV-2. The vaccine, which has been tested in mice, consists of a DNA scaffold that carries many copies of a viral antigen. This type of vaccine, known as a particulate vaccine, mimics the structure of a virus. Most previous work on particulate vaccines has relied on protein scaffolds, but the proteins used in those vaccines tend to generate an unnecessary immune response that can distract the immune system from the target. In the mouse study, the researchers found that the DNA scaffold does not induce an immune response, allowing the immune system to focus its antibody response on the target antigen. “DNA, we found in this work, does not elicit antibodies that may distract away from the protein of interest,” says Mark Bathe, an MIT professor of biological engineering. “What you can imagine is that your B cells and immune system are being fully trained by that target antigen, and that’s what you want — for your immune system to be laser-focused on the antigen of interest.”
|
|
Scooped by
Dr. Stefan Gruenwald
December 19, 2023 6:43 PM
|
The exponential accumulation of digital data is expected to outstrip magnetic and optical storage media. DNA is an incredibly dense (up to 455 exabytes per gram, 6 orders of magnitude denser than magnetic or optical media) and stable (readable over millennia) digital storage medium ( 1– 3). Storage and retrieval of up to gigabytes of digital information in the form of text, images, and movies have been successfully demonstrated. DNA’s essential biological role ensures that the technology for manipulating DNA will never succumb to obsolescence. However, performing computation on the stored data typically involves sequencing the DNA, electronically computing the desired transformation, and synthesizing new DNA. This expensive and slow loop limits the applicability of DNA storage to rarely accessed data (cold storage). In contrast to traditional (passive) DNA storage, schemes for dynamic DNA storage allow access and modification of data without sequencing and/or synthesis. Upon binding to molecular probes, files can be accessed selectively ( 4) and modified through PCR amplification ( 5). Introducing or inhibiting binding of molecular probes with existing data barcodes can rename or delete files ( 6). Information encoded in the hybridization pattern of DNA can be written and erased ( 7) and can even undergo basic logic operations such as AND and OR ( 8) using strand displacement. By encoding information in the nicks of naturally occurring DNA [a.k.a. native DNA ( 9)], data can be modified through ligation or cleavage ( 10). With image similarities encoded in the binding affinities of DNA query probes and data, similarity searches on databases can be performed through DNA hybridization ( 11, 12). Although these advances allow information to be directly accessed and edited within the storage medium, they nevertheless demonstrate limited or no capacity for computation in DNA. Conveniently, beyond its role as a storage medium, DNA has proved to be a programmable medium for computation, primarily via “strand displacement” reactions. With the understanding of the kinetics and thermodynamics of DNA strand displacement ( 13– 15), a variety of rationally designed molecular computing devices have been engineered. These include molecular implementations of logic circuits ( 16– 18), neural networks ( 19, 20), consensus algorithms ( 21), dynamical systems including oscillators ( 22), and cargo-sorting robots ( 23). Given the achievements of strand displacement systems and their inherent molecular parallelism, DNA computation schemes appear well suited to directly carry out computation on big data stored in DNA. A research team now proposes a paradigm called SIMD ||||DNA * (Single Instruction Multiple Data † computation with DNA) which integrates DNA storage with in-memory computation by strand displacement. A preliminary version of the theoretical results appeared as a conference paper ( 25). Inspired by methods of storing information in the positions of nicks in double-stranded DNA ( 9, 10), SIMD ||||DNA encodes information in a multi-stranded DNA complex with a unique pattern of nicks and exposed single-stranded regions (a register). Although storage density is somewhat reduced (approximately a factor of 30), encoding information in nicks still achieves orders of magnitude higher density than magnetic and optical technologies. To manipulate information, an instruction (a set of strands) is applied in parallel to all registers which all share the same sequence space. The strand composition of a register changes when the applied instruction strands trigger strand displacement reactions within that register. Non-reacted instruction strands and reaction waste products are washed away via magnetic bead separation to prepare for the next instruction. Each instruction can change every bit on every register, yielding a high level of parallelism. These experiments routinely manipulated 10^101010 registers in parallel. DNA storage recovery studies suggest that, in principle, 10^8108 distinct register values can be stored in one such sample.
|
|
Rescooped by
Dr. Stefan Gruenwald
from Virus World
November 9, 2023 12:44 PM
|
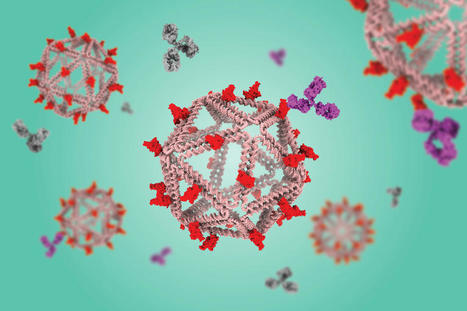
No one had ever seen one virus latching onto another virus, until anomalous sequencing results sent a UMBC team down a rabbit hole leading to a first-of-its-kind discovery. It's known that some viruses, called satellites, depend not only on their host organism to complete their life cycle, but also on another virus, known as a "helper," explains Ivan Erill, professor of biological sciences. The satellite virus needs the helper either to build its capsid, a protective shell that encloses the virus's genetic material, or to help it replicate its DNA. These viral relationships require the satellite and the helper to be in proximity to each other at least temporarily, but there were no known cases of a satellite actually attaching itself to a helper—until now. In a paper published in The ISME Journal, a UMBC team and colleagues from Washington University in St. Louis (WashU) describe the first observation of a satellite bacteriophage (a virus that infects bacterial cells) consistently attaching to a helper bacteriophage at its "neck"—where the capsid joins the tail of the virus. In detailed electron microscopy images taken by Tagide deCarvalho, assistant director of the College of Natural and Mathematical Sciences Core Facilities and first author on the new paper, 80 percent (40 out of 50) helpers had a satellite bound at the neck. Some of those that did not had remnant satellite tendrils present at the neck. Erill, senior author on the paper, describes them as appearing like "bite marks." "When I saw it, I was like, I can't believe this," deCarvalho says. "No one has ever seen a bacteriophage—or any other virus—attach to another virus." A long-term virus relationship After the initial observations, Elia Mascolo, a graduate student in Erill 's research group and co-first author on the paper, analyzed the genomes of the satellite, helper, and host, which revealed further clues about this never-before-seen viral relationship. Most satellite viruses contain a gene that allows them to integrate into the host cell's genetic material after they enter the cell. This allows the satellite to reproduce whenever a helper happens to enter the cell from then on. The host cell also copies the satellite's DNA along with its own when it divides. A bacteriophage sample from WashU also contained a helper and a satellite. The WashU satellite has a gene for integration and does not directly attach to its helper, similar to previously observed satellite-helper systems. However, the satellite in UMBC's sample, named MiniFlayer by the students who isolated it, is the first known case of a satellite with no gene for integration. Because it can't integrate into the host cell's DNA, it must be near its helper—named MindFlayer—every time it enters a host cell if it is going to survive. Given that, although the team did not directly prove this explanation, "attaching now made total sense," Erill says, "because otherwise, how are you going to guarantee that you are going to enter into the cell at the same time?" Additional bioinformatics analysis by Mascolo and Julia López-Pérez, another Ph.D. student working with Erill, revealed that MindFlayer and MiniFlayer have been co-evolving for a long time. "This satellite has been tuning in and optimizing its genome to be associated with the helper for, I would say, at least 100 million years," Erill says, which suggests there may be many more cases of this kind of relationship waiting to be discovered. Research is published in ISME (October 31, 2023): https://doi.org/10.1038/s41396-023-01548-0
Via Juan Lama
|
|
Scooped by
Dr. Stefan Gruenwald
October 10, 2023 8:40 PM
|
A virtual cell modeling system, powered by AI, will lead to breakthroughs in our understanding of diseases, argue the cofounders of the Chan Zuckerberg Initiative. As the smallest living units, cells are key to understanding disease—and yet so much about them remains unknown. We do not know, for example, how billions of biomolecules—like DNA, proteins, and lipids—come together to act as one cell. Nor do we know how our many types of cells interact within our bodies. We have limited understanding of how cells, tissues, and organs become diseased and what it takes for them to be healthy. AI can help us answer these questions and apply that knowledge to improve health and well-being worldwide—if researchers can access and harness these powerful new technologies. Imagine if we had a way to represent every cell state and cell type using AI models. A “virtual cell” could simulate the appearance and known characteristics of any cell type in our body—from the rods and cones that detect light in our retinas to the cardiomyocytes that keep our hearts beating. Scientists could use such a simulator to predict how cells might respond to specific conditions and stimuli: how an immune cell responds to an infection, what happens at the cellular level when a child is born with a rare disease, or even how a patient’s body will respond to a new medication. Scientific discovery, patient diagnosis, and treatment decisions would all become faster, safer, and more efficient. At the Chan Zuckerberg Initiative, we’re helping to generate the scientific data and build out the computing infrastructure to make this a reality—and give scientists the tools they need to take advantage of new advances in AI to help end disease. Advances in AI coupled with large volumes of scientific data have already predicted the structure of nearly all known proteins. DeepMind trained AlphaFold on 50 years’ worth of carefully collected data, and in just five years, they solved the mystery of protein structure. ESM, another AI system which was developed at Meta, is a protein language model trained not on words but on over 60 million protein sequences. It is used for a wide range of applications, like predicting protein structures and the effects of mutations from single sequences. A virtual cell modeling system will also require large amounts of data. Since 2016, CZI has supported researchers globally in efforts to generate and annotate data about cells and their components, built tools to integrate these large data sets, and made them widely available for researchers to learn from and build upon. A global consortium of researchers has been building a reference map of every cell type in the body, and our San Francisco Biohub is creating whole-organism cell atlases. Together, these data sets are yielding the first draft of the open-source Human Cell Atlas, which will chart cell types in the body from development to adulthood. Our SF Biohub and the Chan Zuckerberg Imaging Institute are partnering on OpenCell, which maps the locations of different proteins in our cells. Eric Schmidt: This is how AI will transform the way science gets done Science is about to become much more exciting—and that will affect us all, argues Google's former CEO. Researchers are also using machine-learning models like Geneformer and scGPT to explore large amounts of data about genes and cells—including data generated from CELLxGENE, the open-source software platform that CZI’s science and technology teams created to speed up single-cell research. Similarly, with a new prototype data portal for cryo-electron tomography, our Imaging Institute and our science and technology teams are engaging machine-learning experts to develop automated annotations of microscopy data. This will speed up data processing time from months or even years to just weeks. We are making the data as representative as possible to make sure scientific breakthroughs benefit everyone. This includes incorporating pediatric data into the Human Cell Atlas, filling gaps in our knowledge about the cellular mechanisms of diseases that arise in childhood. With our Ancestry Networks grants, we are also supporting researchers generating reference data about cells based on tissue samples from Black, Latino, Southeast Asian, and Indigenous people, among others from understudied racial, ethnic, and ancestral backgrounds. Already, research teams have made discoveries using these well-curated data sets. One discovered that the broken gene linked to cystic fibrosis is expressed by a type of cell scientists had never come across before, while another identified the respiratory cells that are most vulnerable to SARS-CoV-2. Others are using the data to discover new options for splicing genes to potentially correct disease-causing mutations in specific cells. These discoveries are the first step in developing treatments for diseases—and we believe that AI can significantly speed up researchers’ rate of discoveries going forward. How to compute a virtual cell? To create a virtual cell, we’re building a high-performance computing cluster with 1000+ H100 GPUs that will enable us to develop new AI models trained on various large data sets about cells and biomolecules—including those generated by our scientific institutes. Over time, we hope, this will enable scientists to simulate every cell type in both healthy and diseased states, and query those simulations to see how elusive biological phenomena likely play out—including how cells come into being, how they interact across the body, and how exactly disease-causing changes affect them. The computing cluster won’t be as large as those used in the private sector for commercial products, but once it’s up and running, it will be one of the world’s largest AI clusters for nonprofit scientific research. This will be an important resource for academic teams that are ready to use data sets in new ways but are held back by the prohibitive cost of accessing the latest AI technology. Like our other tools, these digital cell models, and their associated data and applications, will be openly accessible to researchers worldwide.
|
|
Rescooped by
Dr. Stefan Gruenwald
from Virus World
September 15, 2023 7:45 PM
|
Researchers discovered a potential link between "endogenous retroviruses" present in the human genome and the development of neurodegenerative diseases. Summary: Researchers discovered a potential link between “endogenous retroviruses” present in the human genome and the development of neurodegenerative diseases. Their study found that these ancient viral remnants might influence the spread of protein aggregates commonly associated with certain dementias. While these retroviruses don’t trigger neurodegeneration, they may exacerbate the disease process. This discovery offers new potential therapeutic avenues, such as suppressing gene expression or neutralizing viral proteins. Key Facts: - “Endogenous retroviruses”, ancient viral remnants in human DNA, might contribute to neurodegenerative disease progression.
- HERV-W and HERV-K, two such retroviruses, were found to aid in the transport of tau aggregates, protein clumps associated with diseases like Alzheimer’s.
- Potential therapeutic approaches include suppressing the retroviruses or neutralizing their proteins, possibly with antibodies.
Source: DZNE Genetic remnants of viruses that are naturally present in the human genome could affect the development of neurodegenerative diseases. Researchers at DZNE come to this conclusion on the basis of studies on cell cultures. They report on this in the journal Nature Communications. In their view, such “endogenous retroviruses” could contribute to the spread of aberrant protein aggregates – hallmarks of certain dementias – in the brain. Thus, these viral relicts would be potential targets for therapies. It has been suspected for some time that viral infections contribute to the genesis and development of neurodegenerative diseases. Laboratory studies by DZNE scientists now suggest a mechanism that, although related to viruses, does not require infection by external pathogens. According to this study, the culprits would be “endogenous retroviruses” that are naturally present in the human genome. “During evolution, genes from numerous viruses have accumulated in our DNA. Most of these gene sequences are mutated and normally muted,” explained Ina Vorberg, research group leader at DZNE and a professor at the University of Bonn. “However, there is evidence that endogenous retroviruses are activated under certain conditions and contribute to cancer and neurodegenerative diseases. Indeed, proteins or other gene products derived from such retroviruses are found in the blood or tissue of patients.” Experiments with Tau Aggregates Vorberg followed this trail together with colleagues from Bonn and Munich. Using cell cultures, the researchers simulated the situation in which human cells produce certain proteins from the envelope of endogenous retroviruses. Specifically, this involved HERV-W and HERV K – both viruses are present in the human genome but are usually dormant. However, studies indicate that HERV-W is activated in multiple sclerosis and HERV-K in the neurological disease “amyotrophic lateral sclerosis” (ALS) and in frontotemporal dementia (FTD). Now, Vorberg’s team found that the viral proteins facilitate the transport of so-called tau aggregates from cell to cell. “Tau aggregates” are tiny protein clumps that occur in the brains of people affected by certain neurodegenerative diseases – these include Alzheimer’s disease and FTD. “Certainly, conditions in the brain are much more complex than our cellular model system can replicate them. Nevertheless, our experiments show that endogenous retroviruses can influence the spread of tau aggregates between cells,” Vorberg said. “Endogenous retroviruses would thus not be triggers of neurodegeneration, but could fuel the disease process once it is already underway.” Viral Transport Mediators The current research and earlier studies by Vorberg’s team suggest that viral proteins serve as transport mediators for tau aggregates because they insert into the cell membrane and into the membrane of so-called extracellular vesicles: These are small fat bubbles that are naturally secreted by cells. “For the transport of tau aggregates from cell to cell, we see two pathways in particular. Transfer between cells that are in direct contact, and transport within vesicles that act as cargo capsules, so to speak, and pass from one cell to another to eventually merge with it,” Vorberg explained. “In both scenarios, membranes have to fuse. Proteins from the envelope of viruses can promote this process. That’s because many viruses are adapted to fuse with host cells. “This happens by means of special proteins that viruses carry on their surfaces. If precisely these proteins are incorporated into the cell membrane and the membrane of extracellular vesicles, it is understandable that the tau aggregates then spread more easily.” Starting Points for Therapy In the course of the natural aging process, the regulation of genes can change – originally “dormant” endogenous retroviruses could be “awakened” as a result. Indeed, the symptoms of most neurodegenerative diseases do not manifest until older age. This raises two conceivable approaches to therapy. “On the one hand, one could try to specifically suppress gene expression, that is, to inactivate the endogenous retroviruses again. That would get to the root of the problem,” Vorberg said. “But you could also start elsewhere and try to neutralize the viral proteins – for example, with antibodies.” Searching for Antibodies In the opinion of the researchers, it is likely that dementia patients with tau aggregates carry increased amounts of such antibodies. If it were possible to isolate these and reproduce them using biotechnological methods, it might be possible to develop a passive vaccine. Thus, in collaboration with DZNE colleagues in Berlin and Bonn, Vorberg’s team aims to specifically search for such antibodies in patients. In addition, the scientists are considering antiviral drugs. In cell culture, they have already found that such agents can actually stop the spread of protein aggregates. “This is another approach we intend to pursue,” said Vorberg. Research cited published in Nature (Aug. 18, 2023): https://doi.org/10.1038/s41467-023-40632-z
Via Juan Lama
|
|
Scooped by
Dr. Stefan Gruenwald
July 7, 2023 6:52 PM
|
Creating new drug candidates is a heroic endeavor, often taking over 10 years to bring a drug to market. New supercomputing-scale large language models (LLMs) that understand biology and chemistry text are helping scientists understand proteins, small molecules, DNA, and biomedical text. These state-of-the-art AI models help generate de novo proteins and molecules and predict the 3D structures of proteins. They can predict the binding structure of a small molecule to a protein and are offering scientists easier ways to engineer new candidate drugs and ultimately bring hope for patients. After Exscientia brought an AI-designed drug candidate to a clinical trial in 2021, several other companies have announced that their candidates are in trials. Within drug companies focused on AI-based discovery, there is publicly available information on about 160 discovery programs, of which 15 products are reportedly in clinical development. At the forefront of AI-based drug discovery are generative AI models for applications such as generating high-quality proteins. These large, powerful models learn from unlabeled data (such as sequencing data) on multi-GPU, multi-node, high-performance computing (HPC) infrastructures. With the novel NVIDIA BioNeMo Service, workflows for generative AI for biology are optimized and turnkey. You can focus on adapting AI models to the right drug candidates instead of dealing with configuration files and setting up supercomputing infrastructure. BioNeMo Service BioNeMo Service is a cloud service for generative AI in early drug discovery, featuring nine state-of-the-art large language and diffusion models in one place. The models in BioNeMo are accessible through a web interface or fully managed APIs and can be further trained and optimized on NVIDIA DGX Cloud. With BioNeMo Service, you can perform any of the following tasks: - Generate large libraries of proteins.
- Build property predictors using embeddings to refine protein libraries.
- Generate small molecules with specific properties.
- Rapidly and accurately predict and visualize the 3D structure for billions of proteins.
- Run large campaigns of ligand-to-small-molecule pose estimations.
- Download proteins, molecules, and predicted 3D structures.
Generative AI models in BioNeMo Service BioNeMo Service features nine AI generative models covering a wide spectrum of applications for developing AI drug discovery pipelines: - AlphaFold 2, ESMFold, and OpenFold for 3D protein structure prediction from a primary amino acid sequence
- ESM-1nv and ESM-2 for protein property predictions
- ProtGPT2 for protein generation
- MegaMolBART and MoFlow for small molecule generation
- DiffDock for predicting the binding structure of a small molecule to a protein
|
|
Rescooped by
Dr. Stefan Gruenwald
from Virus World
June 18, 2023 11:52 AM
|
Researchers recently discovered an unexpected link between the progression of ALS and PEG10, a protein traditionally known for its role in placental development. Overabundance of this protein in nerve tissue has been observed to alter cell behavior contributing to ALS. The team is now studying the molecular pathways involved with a view to inhibiting this rogue protein, potentially paving the way for new therapeutics. Key facts: - The ancient, virus-like protein PEG10, typically associated with placental development, has been linked to the progression of ALS when present in high levels in nerve tissue.
- The research marks the first time PEG10 has been connected to ALS, with its accumulation seen as a hallmark of the disease.
- Research findings suggest that inhibiting the rogue PEG10 protein could potentially lead to a new class of therapeutics for ALS.
More than 5,000 people are diagnosed annually with ALS (amyotrophic lateral sclerosis), a fatal, neurodegenerative disease that attacks nerve cells in the brain and spinal cord, gradually robbing people of the ability to speak, move, eat and breathe. To date, only a handful of drugs exist to moderately slow its progression. There is no cure. But CU Boulder researchers have identified a surprising new player in the disease—an ancient, virus-like protein that is best known, paradoxically, for its essential role in enabling placental development. The findings were recently published in the journal eLife. “Our work suggests that when this strange protein known as PEG10 is present at high levels in nerve tissue, it changes cell behavior in ways that contribute to ALS,” said senior author Alexandra Whiteley, assistant professor in the Department of Biochemistry. Whiteley’s lab is now working to understand the molecular pathways involved and to find a way of inhibiting the rogue protein. “It is early days still, but the hope is this could potentially lead to an entirely new class of potential therapeutics to get at the root cause of this disease.” Ancient viruses with modern-day impact Mounting research suggests about half the human genome is made up of bits of DNA left behind by viruses (known as retroviruses) and similar virus-like parasites, known as transposons, which infected our primate ancestors 30-50 million years ago. Some, like HIV, are well known for their ability to infect new cells and cause disease. Others, like wolves who have lost their fangs, have become domesticated over time, losing their ability to replicate while continuing to pass from generation to generation, shaping human evolution and health. PEG10, or Paternally Expressed Gene 10, is one such “domesticated retrotransposon.” Studies show it likely played a key role in enabling mammals to develop placentas—a critical step in human evolution. But like a viral Jekyll and Hyde, when it’s overly abundant in the wrong places, it may also fuel disease, including certain cancers and another rare neurological disorder called Angelman’s syndrome, studies suggest. Whiteley’s research is the first to link the virus-like protein to ALS, showing that PEG10 is present in high levels in the spinal cord tissue of ALS patients where it likely interferes with the machinery enabling brain and nerve cells to communicate. “It appears that PEG10 accumulation is a hallmark of ALS,” said Whiteley, who has already secured a patent for PEG10 as a biomarker, or way of diagnosing, the disease. Too much protein in the wrong places Whiteley did not set out to study ALS, or ancient viruses. Instead, she studies how cells get rid of extra protein, as too much of the typically good thing has been implicated in other neurodegenerative diseases, including Alzheimer’s and Parkinson’s. Her lab is one of a half-dozen in the world to study a class of genes called ubiquilins, which serve to keep problem proteins from accumulating in cells. In 2011, a study linked a mutation in the ubiquilin-2 gene (UBQLN2) to some cases of familial ALS, which makes up about 10% of ALS cases. The other 90% are sporadic, meaning they are not believed to be inherited. But it has remained unclear how the faulty gene might fuel the deadly disease. Using laboratory techniques and animal models, Whiteley and colleagues at Harvard Medical School first set out to determine which proteins pile up when the UBQLN2 misfires and fails to put the brakes on. Among thousands of possible proteins, PEG10 topped the list. Then Whiteley and her colleagues collected the spinal tissue of deceased ALS patients (provided by the medical research foundation Target ALS) and used protein analysis, or proteomics, to see which if any seemed overexpressed. Again, among more than 7,000 possible proteins, PEG10 was in the top five. In a separate experiment, the team found that with the ubiquilin brakes essentially broken, the PEG10 protein piles up and disrupts the development of axons—the cords which carry electrical signals from the brain to the body. PEG10 was over-expressed in the tissue of individuals with both sporadic and familial ALS, the study found, meaning the virus-like protein may be playing a key role in both. “The fact that PEG10 is likely contributing to this disease means we may have a new target for treating ALS,” she said. “For a terrible disease in which there are no effective therapeutics that lengthen lifespan more than a couple of months, that could be huge.” The research could also lead to a better understanding of other diseases that result from protein accumulation, as well as keener insight into how ancient viruses influence health. In this case, Whiteley said, the so-called “domesticated” virus could a be rearing its fangs again. “Domesticated is a relative term, as these virus-like activities may be a driver of neurodegenerative disease,” she said. “And in this case, what is good for the placenta may be bad for neural tissue.” Original research published in eLIfe(May 23, 2023): https://doi.org/10.7554/eLife.79452
Via Juan Lama
|
|
Scooped by
Dr. Stefan Gruenwald
May 27, 2023 11:21 AM
|
For more than a century, biologists have wondered what the earliest animals were like when they first arose in the ancient oceans over half a billion years ago. Searching among today’s most primitive-looking animals for the earliest branch of the animal tree of life, scientists gradually narrowed the possibilities down to two groups: sponges, which spend their entire adult lives in one spot, filtering food from seawater; and comb jellies, voracious predators that oar their way through the world’s oceans in search of food. In a new study published this week in the journal Nature, researchers use a novel approach based on chromosome structure to come up with a definitive answer: Comb jellies, or ctenophores (teen’-a-fores), were the first lineage to branch off from the animal tree. Sponges were next, followed by the diversification of all other animals, including the lineage leading to humans. Although the researchers determined that the ctenophore lineage branched off before sponges, both groups of animals have continued to evolve from their common ancestor. Nevertheless, evolutionary biologists believe that these groups still share characteristics with the earliest animals, and that studying these early branches of the animal tree of life can shed light on how animals arose and evolved to the diversity of species we see around us today. “The most recent common ancestor of all animals probably lived 600 or 700 million years ago. It’s hard to know what they were like because they were soft-bodied animals and didn’t leave a direct fossil record. But we can use comparisons across living animals to learn about our common ancestors,” said Daniel Rokhsar, University of California, Berkeley professor of molecular and cell biology and co-corresponding author of the paper along with Darrin Schultz and Oleg Simakov of the University of Vienna. “It’s exciting — we’re looking back deep in time where we have no hope of getting fossils, but by comparing genomes, we’re learning things about these very early ancestors.” Understanding the relationships among animal lineages will help scientists understand how key features of animal biology, such as the nervous system, muscles and digestive tract, evolved over time, the researchers say. “We developed a new way to take one of the deepest glimpses possible into the origins of animal life,” said Schultz, the lead author and a former UC Santa Cruz graduate student and researcher at the Monterey Bay Aquarium Research Institute (MBARI) who is now a postdoctoral researcher at the University of Vienna. “This finding will lay the foundation for the scientific community to begin to develop a better understanding of how animals have evolved.” What is an animal?
Most familiar animals, including worms, flies, mollusks, sea stars and vertebrates — and including humans — have a head with a centralized brain, a gut running from mouth to anus, muscles and other shared features that had already evolved by the time of the famed “Cambrian Explosion” around 500 million years ago. Together, these animals are called bilaterians. Other bona fide animals, however, such as jellyfish, sea anemones, sponges and ctenophores, have simpler body plans. These creatures lack many bilaterian features — for example, they lack a defined brain and may not even have a nervous system or muscles — but still share the hallmarks of animal life, notably the development of multicellular bodies from a fertilized egg. The evolutionary relationships among these diverse creatures — specifically, the order in which each of the lineages branched off from the main trunk of the animal tree of life — has been controversial. With the rise of DNA sequencing, biologists were able to compare the sequences of genes shared by animals to construct a family tree that illustrates how animals and their genes evolved over time since the earliest animals arose in the Precambrian Period. But these phylogenetic methods based on gene sequences failed to resolve the controversy over whether sponges or comb jellies were the earliest branch of the animal tree, in part because of the deep antiquity of their divergence, Rokhsar said. “The results of sophisticated sequence-based studies were basically split,” he said. “Some researchers did well-designed analyses and found that sponges branched first. Others did equally complex and justifiable studies and got ctenophores. There hasn’t really been any convergence to a definitive answer.”
|
|
Scooped by
Dr. Stefan Gruenwald
May 17, 2023 6:24 PM
|
An international research team led by the Max Planck Institute for Evolutionary Anthropology in Leipzig, Germany, has for the first time successfully isolated ancient human DNA from a Paleolithic artefact: a pierced deer tooth discovered in Denisova Cave in southern Siberia. To preserve the integrity of the artefact, they developed a new, nondestructive method for isolating DNA from ancient bones and teeth. From the DNA retrieved they were able to reconstruct a precise genetic profile of the woman who used or wore the pendant, as well as of the deer from which the tooth was taken. Genetic dates obtained for the DNA from both the woman and the deer show that the pendant was made between 19,000 and 25,000 years ago. The tooth remains fully intact after analysis, providing testimony to a new era in ancient DNA research, in which it may become possible to directly identify the users of ornaments and tools produced in the deep past. Pierced deer tooth discovered from Denisova Cave in southern Siberia that yielded ancient human DNA. Artefacts made of stone, bones or teeth provide important insights into the subsistence strategies of early humans, their behavior and culture. However, until now it has been difficult to attribute these artefacts to specific individuals, since burials and grave goods were very rare in the Palaeolithic. This has limited the possibilities of drawing conclusions about, for example, division of labor or the social roles of individuals during this period. In order to directly link cultural objects to specific individuals and thus gain deeper insights into Paleolithic societies, an international, interdisciplinary research team, led by the Max Planck Institute for Evolutionary Anthropology in Leipzig, has developed a novel, non-destructive method for DNA isolation from bones and teeth. Although they are generally rarer than stone tools, the scientists focused specifically on artefacts made from skeletal elements, because these are more porous and are therefore more likely to retain DNA present in skin cells, sweat and other body fluids. A new DNA extraction method Lead author Elena Essel working in the clean laboratory on the pierced deer tooth discovered from Denisova Cave. Before the team could work with real artefacts, they first had to ensure that the precious objects would not be damaged. “The surface structure of Paleolithic bone and tooth artefacts provides important information about their production and use. Therefore, preserving the integrity of the artefacts, including microstructures on their surface, was a top priority” says Marie Soressi, an archaeologist from the University of Leiden who supervised the work together with Matthias Meyer, a Max Planck geneticist. The team tested the influence of various chemicals on the surface structure of archaeological bone and tooth pieces and developed a non-destructive phosphate-based method for DNA extraction. “One could say we have created a washing machine for ancient artifacts within our clean laboratory," explains Elena Essel, the lead author of the study who developed the method. "By washing the artifacts at temperatures of up to 90°C, we are able to extract DNA from the wash waters, while keeping the artifacts intact.”
|
|
Scooped by
Dr. Stefan Gruenwald
May 14, 2023 2:04 PM
|
Humans inherited genetic material from Neanderthals that affects the shape of our noses, finds a new study led by UCL researchers. The new Communications Biology study finds that a particular gene, which leads to a taller nose (from top to bottom), may have been the product of natural selection as ancient humans adapted to colder climates after leaving Africa. Co-corresponding author Dr Kaustubh Adhikari (UCL Genetics, Evolution & Environment and The Open University) said: "In the last 15 years, since the Neanderthal genome has been sequenced, we have been able to learn that our own ancestors apparently interbred with Neanderthals, leaving us with little bits of their DNA. "Here, we find that some DNA inherited from Neanderthals influences the shape of our faces. This could have been helpful to our ancestors, as it has been passed down for thousands of generations." The study used data from more than 6,000 volunteers across Latin America, of mixed European, Native American and African ancestry, who are part of the UCL-led CANDELA study, which recruited from Brazil, Colombia, Chile, Mexico and Peru. The researchers compared genetic information from the participants to photographs of their faces -- specifically looking at distances between points on their faces, such as the tip of the nose or the edge of the lips -- to see how different facial traits were associated with the presence of different genetic markers. The researchers newly identified 33 genome regions associated with face shape, 26 of which they were able to replicate in comparisons with data from other ethnicities using participants in east Asia, Europe, or Africa. In one genome region in particular, called ATF3, the researchers found that many people in their study with Native American ancestry (as well as others with east Asian ancestry from another cohort) had genetic material in this gene that was inherited from the Neanderthals, contributing to increased nasal height. They also found that this gene region has signs of natural selection, suggesting that it conferred an advantage for those carrying the genetic material. First author Dr Qing Li (Fudan University) said: "It has long been speculated that the shape of our noses is determined by natural selection; as our noses can help us to regulate the temperature and humidity of the air we breathe in, different shaped noses may be better suited to different climates that our ancestors lived in. The gene we have identified here may have been inherited from Neanderthals to help humans adapt to colder climates as our ancestors moved out of Africa." Co-author Prof. Andres Ruiz-Linares (Fudan University, UCL Genetics, Evolution & Environment, and Aix-Marseille University) added: "Most genetic studies of human diversity have investigated the genes of Europeans; our study's diverse sample of Latin American participants broadens the reach of genetic study findings, helping us to better understand the genetics of all humans." The finding is the second discovery of DNA from archaic humans, distinct from Homo sapiens, affecting our face shape. The same team discovered in a 2021 paper that a gene influencing lip shape was inherited from the ancient Denisovans.*
|
|
Rescooped by
Dr. Stefan Gruenwald
from Virus World
May 1, 2023 12:08 PM
|
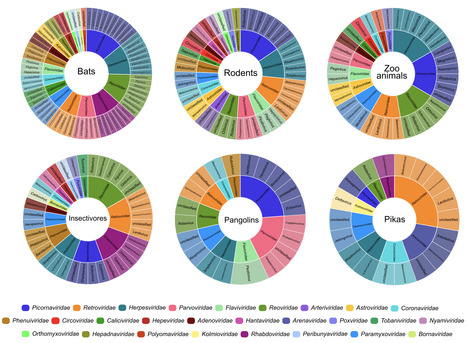
Wildlife is reservoir of emerging viruses. Here we identified 27 families of mammalian viruses from 1981 wild animals and 194 zoo animals collected from south China between 2015 and 2022, isolated and characterized the pathogenicity of eight viruses. Bats harbor high diversity of coronaviruses, picornaviruses and astroviruses, and a potentially novel genus of Bornaviridae. In addition to the reported SARSr-CoV-2 and HKU4-CoV-like viruses, picornavirus and respiroviruses also likely circulate between bats and pangolins. Pikas harbor a new clade of Embecovirus and a new genus of arenaviruses. Further, the potential cross-species transmission of RNA viruses (paramyxovirus and astrovirus) and DNA viruses (pseudorabies virus, porcine circovirus 2, porcine circovirus 3 and parvovirus) between wildlife and domestic animals was identified, complicating wildlife protection and the prevention and control of these diseases in domestic animals. This study provides a nuanced view of the frequency of host-jumping events, as well as assessments of zoonotic risk. Monitoring the diversity of viruses infecting animals is important for assessing zoonotic risk. Here, the authors use metatranscriptomics to characterise the viromes of small mammals, pangolins, and zoo animals in China to identify potentially zoonotic viruses. Published in Nature Comm. (April 29, 2023): https://doi.org/10.1038/s41467-023-38202-4
Via Juan Lama
|
|
Scooped by
Dr. Stefan Gruenwald
March 31, 2023 12:45 PM
|
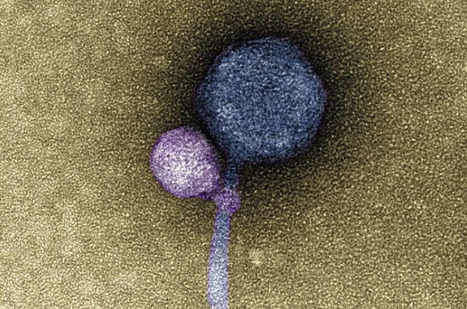
P. asymbiotica, a bacteria that normally grows inside C.elegans roundworms, uses these nematodes as Trojan horses to invade insect larvae. It works like this: a nematode invades the larva's body and regurgitates P. asymbiotica; the bacteria kills the insect's cells; and the nematode feasts on the dying larva's flesh. Thus, the nematodes and bacteria enjoy a beautiful symbiotic relationship. To kill the insect cells, P. asymbiotica secretes tiny, spring-loaded syringes, scientifically known as "extracellular contractile injection systems," that carry toxic proteins inside a hollow "needle" with a spike on one end. Small "tails" extend from the base of the syringe — imagine the landing gear of a space probe — and these tails bind to proteins on the surface of insect cells. Once bound, the syringe stabs its needle through the cell membrane to release its cargo. In previous studies, scientists isolated these syringes from Photorhabdus bacteria and also discovered that some could target mouse cells, not just insect cells. This raised the possibility that such syringes could be modified for use in humans. To test whether this idea might be feasible, the team first loaded the syringe's hollow tube with proteins of their choosing. Then, they used AlphaFold to better understand how the syringes hone in on insect cells, so they could be modified to target human cells instead. They used the AI system to predict the structure of the bottom of the syringe's landing gear — the part that first makes contact with the target cell surface. They then altered this structure so it would latch onto surface proteins found only on human cells. Without Deepmind's AlphaFold, the researchers would have had to conduct this analysis using advanced microscopy techniques and crystallography, meaning detailed studies of the landing gear's atomic structure, Joseph Kreitz, a doctoral student at the McGovern Institute for Brain Research at MIT and first author of the study, told Live Science in an email. "This could have taken many months," Kreitz said. "With AlphaFold, we were able to obtain predicted structures of candidate tail fiber designs almost in real-time, significantly accelerating our efforts to reprogram this protein." The researchers then used their modified syringes to tweak cells' genomes in lab dishes. Specifically, they delivered components of the powerful CRISPR-Cas9 gene editing tool into cells to cut and paste sections of DNA into their genomes. The team also used the syringes to insert tiny DNA-snipping scissors called zinc-finger deaminases into cells. They also used the system to deliver toxic proteins into cancer cells in lab dishes. And finally, they injected the syringes into live mice and found that their cargo could only be detected in the targeted areas and did not spark a harmful immune reaction. For this last experiment, the team used AlphaFold to design their syringes to specifically target mouse cells. These experiments demonstrate that the syringes can serve as "programmable protein delivery devices with possible applications in gene therapy, cancer therapy and biocontrol," the authors concluded. In contrast to therapies that deliver genetic instructions, like DNA or RNA, into cells, these protein-carrying syringes could provide "better control over the dose and half-life of a therapeutic inside cells," Kreitz and the study's senior author Feng Zhang told Live Science in an email. That's because genetic instructions prompt cells to build proteins for themselves, whereas the syringes would come with a premeasured dose of protein. This precise dosing would be useful for treatments involving transcription factors, which tweak a cell's gene activity, and chemotherapy, which has toxic effects at high doses, they said. The tiny syringes could also potentially be programmed to fight disease-causing bacteria in the body, Ericson and Pilhofer wrote. And in the future, it may be possible for scientists to connect multiple syringes to form multi-barreled complexes. "These might enable more cargo to be delivered per target cell than with a single injection system," they suggested. "However, we note that this system is still in its infancy; further efforts will be required to characterize the behavior of this system in vivo before it can be applied in clinical or commercial settings," Kreitz and Zhang told Live Science. The team is now studying how well the syringes diffuse through different tissues and organs, and continuing to examine how the immune system reacts to the new protein delivery system.
|
|
Rescooped by
Dr. Stefan Gruenwald
from TAL effector science
March 28, 2023 8:43 PM
|
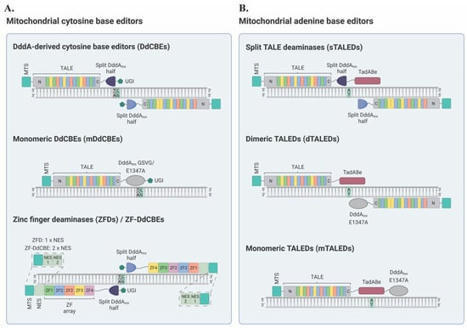
Mitochondria are critical organelles that form networks within our cells, generate energy dynamically, contribute to diverse cell and organ function, and produce a variety of critical signaling molecules, such as cortisol. This intracellular microbiome can differ between cells, tissues, and organs. Mitochondria can change with disease, age, and in response to the environment. Single nucleotide variants in the circular genomes of human mitochondrial DNA are associated with many different life-threatening diseases. Mitochondrial DNA base editing tools have established novel disease models and represent a new possibility toward personalized gene therapies for the treatment of mtDNA-based disorders. Primary mtDNA-based disorders are currently incurable [ 2] and these diseases often cause significant illness and can lead to premature death [ 2]. Moreover, variations in mtDNA can occur in individuals who are otherwise healthy and have been implicated in the etiology of age-related multifactorial diseases, including neurodegenerative disorders such as Parkinson’s disease, metabolic conditions, heart failure, and several forms of cancer [ 10, 15]. The increasing prevalence of these conditions in our aging population highlights the need for the development of novel approaches for the investigation and prevention or treatment of these disorders [ 12, 16]. Gene editing techniques can introduce targeted DNA modifications in cells or tissues to correct a genetic defect, and have already been successfully used to correct pathogenic mutations in the nuclear genome [ 17, 18]. Mitochondrial gene therapy is a relatively new idea that, despite several challenges, has seen significant progress over the last few years [ 19]. Mitochondrial gene editing technologies can be designed to specifically act on variant mtDNA molecules, driving a heteroplasmic state toward a healthy, wild-type mtDNA population [ 20, 21]. Broadly, two distinct modalities are currently used for mtDNA manipulation: (i) nuclease-based and (ii) base editing approaches. Conceptually, nuclease-based methods can be utilized to decrease the amount of variant mtDNA in mitochondria by specifically targeting and cleaving the mutant mtDNA molecules [ 22, 23]. This technique relies on the premise that double-strand breaks (DSBs) in mtDNA induce the rapid degradation of the linearized molecule, instead of its repair [ 20, 23]. If mutant mtDNA is specifically eliminated, the residual mtDNA, mostly wild-type, replicates and repopulates the organelle, resulting in the restoration of normal mtDNA levels. In particular, nuclease-based approaches have been described to include mitochondrially targeted restriction endonucleases (mitoREs), zinc-finger nucleases (mtZFNs), and transcription activator-like effector nucleases (mitoTALENs) [ 20, 22, 23, 24, 25, 26]. These techniques have been extensively reviewed elsewhere [ 27, 28, 29, 30]. Additionally, mitochondrially targeted CRISPR (mitoCRISPR) systems have also been reported [ 31, 32, 33, 34]. However, these mitoCRISPR platforms have yet to be widely accepted within the scientific community due to the challenging nature of guide RNA (gRNA) import into the mitochondrial matrix, as well as a notable lack of follow-up studies [ 27, 35, 36]. Despite their potential usefulness for shifting heteroplasmy, mitochondrically-targeted nucleases are unable to correct variant genomes [ 20, 22]. Therefore, nuclease-based approaches cannot be utilized to rescue pathological conditions in homoplasmic states [ 20, 28]. As mentioned above, the main alternative to nuclease-based strategies for mtDNA manipulation is base editing [ 37, 38]. Consequently, in this work, the scientists focused on recent advancements in the field of mitochondrial base editing, which holds the potential to treat diseases caused by pathogenic mtDNA point mutations in both heteroplasmic and homoplasmic contexts without the risk for mtDNA depletion [ 39, 40]. This review also sought to provide insights into paths toward the development of therapeutic approaches for mtDNA-based disorders and the establishment of mitochondrial disease models to better understand the biology of these devastating diseases.
Via dromius
|
|
|
Scooped by
Dr. Stefan Gruenwald
February 19, 2024 2:56 AM
|
Armed with a catalog of hundreds of thousands of DNA and RNA virus species in the world's oceans, scientists are now zeroing in on the viruses most likely to combat climate change by helping trap carbon dioxide in seawater or, using similar techniques, different viruses that may prevent methane's escape from thawing Arctic soil. By combining genomic sequencing data with artificial intelligence analysis, researchers have identified ocean-based viruses and assessed their genomes to find that they "steal" genes from other microbes or cells that process carbon in the sea. Mapping microbial metabolism genes, including those for underwater carbon metabolism, revealed 340 known metabolic pathways throughout the global oceans. Of these, 128 were also found in the genomes of ocean viruses. "I was shocked that the number was that high," said Matthew Sullivan, professor of microbiology and director of the Center of Microbiome Science at The Ohio State University. Having mined this massive trove of data via advances in computation, the team has now revealed which viruses have a role in carbon metabolism and are using this information in newly developed community metabolic models to help predict how using viruses to engineer the ocean microbiome toward better carbon capture would look. "The modeling is about how viruses may dial up or dial down microbial activity in the system," Sullivan said. "Community metabolic modeling is telling me the dream data point: which viruses are targeting the most important metabolic pathways, and that matters because it means they're good levers to pull on."
|
|
Rescooped by
Dr. Stefan Gruenwald
from Virus World
November 26, 2023 2:11 PM
|
At age 45, Dr. Lakiea Bailey said, she was the oldest person with sickle cell anemia that she knew. The executive director of the nonprofit patient advocacy group the Sickle Cell Consortium was diagnosed with sickle cell disease at age 3. Because of it, she’s had heart problems, had her hips replaced, and experienced serious pain all her life. Bailey told the US Food and Drug Administration’s independent advisory committee that she believes a cutting-edge therapy that is currently under review offers the sickle cell community something many haven’t ever had before: hope. “Hope is on the horizon, and we are looking toward this hope for a change of the lives that we are living of excruciating pain,” Bailey told the FDA committee. The independent committee is helping the FDA think through how it should evaluate a treatment called exa-cel that could potentially cure people of sickle cell disease, a painful and deadly disease with no universally successful treatment. This was an ongoing discussion with no vote or decision about the therapy, but the discussion likely moves the US one step closer to approving a groundbreaking new treatment that uses gene editing. If approved, exa-cel, made by Boston-based Vertex Pharmaceuticals and the Swiss company CRISPR Therapeutics, would be the first FDA-approved treatment that uses genetic modification called CRISPR. CRISPR, or clustered regularly interspaced short palindromic repeats, is a technology researchers use to selectively modify DNA, the carrier of genetic information that the body uses to function and develop. The FDA said treatment for severe sickle cell is an “unmet medical need.” When someone has sickle cell disease their red blood cells don’t function the way they should. Red blood cells are the helper cells that carry oxygen from the lungs to the body’s tissues, which use this oxygen to produce energy. The process also generates waste in the form of carbon dioxide that the red blood cells take to the lungs to be exhaled out. With sickle cell disease — also called sickle cell anemia — red blood cells take on a folded or sickle shape that can clog tiny blood vessels and cause progressive organ damage and pain, and can lead to organ failure. The sickle cells also die earlier than they should, which means the person is constantly short red blood cells. One person with sickle cell who testified said she had been hospitalized 100 times just last year. Median life expectancy is only 45 years. Sickle cell is rare, and it disproportionately impacts African Americans. About 100,000 people in the US are diagnosed with sickle cell and, of those, 20,000 have what’s considered severe disease. Up until now, the only real treatment has been a stem cell or bone marrow transplant. For stem cells, fewer than 20% of patients have an appropriately matched donor, the FDA said, and the transplants are risky and may not work. Sometimes a transplant can kill the patient. The new exa-cel treatment under FDA consideration can use the patient’s own stem cells. Doctors would alter them with CRISPR to fix the genetic problems that cause sickle cell, and then the altered stem cells are given back to the patient in a one-time infusion. In company studies, the treatment was considered safe, and it had a “highly positive benefit-risk for patients with severe sickle cell disease,” Dr. Stephanie Krogmeier, vice president for global regulatory affairs with Vertex Pharmaceuticals Incorporated, told the panel. Thirty-nine of the 40 people tested with the treatment did not have a single vaso-occlusive crisis, which means the misshapen red blood cells block normal circulation and can cause moderate to severe pain. It’s the top reason patients with sickle cell go to the emergency room or are hospitalized. Before the treatment, patients experienced about four of these painful crises a year, resulting in about two weeks in the hospital. The FDA sought the independent panel’s advice, in part, because this would be the first time the FDA would approve a treatment that uses CRISPR technology, but Dr. Fyodor Urnov, a professor in the Department of Molecular and Cell Biology at the University of California, Berkeley, reminded the committee CRISPR has been around for 30 years and, in that time, scientists have learned a lot about how to use it safely. “The technology is, in fact, ready for primetime,” Urnov said. With this kind of genetic editing, scientists could inadvertently make a change to a patient’s DNA that is off-target, and the therapy could harm the patient. The FDA wanted the experts’ advice so it could understand what criteria it should use to evaluate the treatment and determine how to evaluate long-term safety issues. An FDA presentation to the panel suggested the agency may have some questions about the data. It called a lack of confirmatory testing “concerning.” It also noted the study’s small size. In a discussion of the company’s methodology, several panel experts said that they believed the data the companies have submitted for FDA approval were reasonable. Committee member Dr. Gil Wolfe, a distinguished professor in the department of neurology at University at Buffalo Jacobs School of Medicine and Biomedical Sciences, said he liked that the company promised to monitor patients for 15 years to see if there were any problems down the road. He said, generally, it was “exciting” to see how many patients have been treated and how positive the results have been so far. As far as any concern for what’s called “off-target effects,” meaning the potential unwanted or adverse alterations to the genome that could accidentally happen in this process and cause cancer or other problems down the road, Dr. Daniel Bauer, principal investigator and staff physician at Dana-Farber/Boston Children’s Cancer and Blood Disorders Center, Boston Children’s Hospital, told the panel the risk is “relatively small.” Wolfe thought the depth of analysis the companies used should be good enough to detect any potential problems down the road. “We want to be careful to not let the perfect be the enemy of the good,” Wolfe said. “At some point, you have to just try things out.” “I think, in this case, that there’s a huge unmet need for individuals with sickle cell disease, and it’s important we think about how we can advance therapies that could potentially help them, and I certainly think this is one of them,” Wolfe added. Asked by the committee how he would advise patients how to evaluate the risks with this treatment, Bauer said he would be honest that there is some uncertainty, but most of the human genome is non-coding, meaning it doesn’t provide instructions to the cells to act a certain way. “It might be that many places in the human genome can tolerate an off-target edit and not have a functional consequence,” Bauer told the committee. In other words, if they made an editing error, it might not matter, and may not harm the patient. “My guess is it’s a relatively small risk in the scheme of this risk benefit. But it’s new, it’s unknown,” Bauer said. “We need to be humble and open to learning from these brave patients participating.” The FDA is expected to make an approval decision by December 8, 2023.
Via Juan Lama
|
|
Rescooped by
Dr. Stefan Gruenwald
from Virus World
October 11, 2023 1:34 PM
|
A new study highlights both the promise and the limitations of gene editing, as a highly lethal form of avian influenza continues to spread around the world. Scientists have used the gene-editing technology known as CRISPR to create chickens that have some resistance to avian influenza, according to a new study that was published in the journal Nature Communications on Tuesday. The study suggests that genetic engineering could potentially be one tool for reducing the toll of bird flu, a group of viruses that pose grave dangers to both animals and humans. But the study also highlights the limitations and potential risks of the approach, scientists said. Some breakthrough infections still occurred, especially when gene-edited chickens were exposed to very high doses of the virus, the researchers found. And when the scientists edited just one chicken gene, the virus quickly adapted. The findings suggest that creating flu-resistant chickens will require editing multiple genes and that scientists will need to proceed carefully to avoid driving further evolution of the virus, the study’s authors said. The research is “proof of concept that we can move toward making chickens resistant to the virus,” Wendy Barclay, a virologist at Imperial College London and an author of the study, said at a news briefing. “But we’re not there yet.” Some scientists who were not involved in the research had a different takeaway. “It’s an excellent study,” said Dr. Carol Cardona, an expert on bird flu and avian health at the University of Minnesota. But to Dr. Cardona, the results illustrate how difficult it will be to engineer a chicken that can stay a step ahead of the flu, a virus known for its ability to evolve swiftly. “There’s no such thing as an easy button for influenza,” Dr. Cardona said. “It replicates quickly, and it adapts quickly.” What to Know About Avian Flu The spread of H5N1. A new variant of this strain of the avian flu has spread widely through bird populations in recent years. It has taken an unusually heavy toll on wild birds and repeatedly spilled over into mammals, including minks, foxes and bears. Here’s what to know about the virus: What is avian influenza? Better known as the bird flu, avian influenza is a group of flu viruses that is well adapted to birds. Some strains, like the version of H5N1 that is currently spreading, are frequently fatal to chickens and turkeys. It spreads via nasal secretions, saliva and fecal droppings, which experts say makes it difficult to contain. Should humans be worried about being infected? Although the danger to the public is currently low, people who are in close contact with sick birds can and have been infected. The virus is primarily a threat to birds, but infections in mammals increase the odds that the virus could mutate in ways that make it more of a risk to humans, experts say. How can we stop the spread? The U.S. Department of Agriculture has urged poultry growers to tighten their farms’ biosecurity measures, but experts say the virus is so contagious that there is little choice but to cull infected flocks. The Biden administration has been contemplating a mass vaccination campaign for poultry. Is it safe to eat poultry and eggs? The Agriculture Department has said that properly prepared and cooked poultry and eggs should not pose a risk to consumers. The chance of infected poultry entering the food chain is “extremely low,” according to the agency. Can I expect to pay more for poultry products? Egg prices soared when an outbreak ravaged the United States in 2014 and 2015. The current outbreak of the virus — paired with inflation and other factors — has contributed to an egg supply shortage and record-high prices in some parts of the country. Avian influenza refers to a group of flu viruses that are adapted to spread in birds. Over the last several years, a highly lethal version of a bird flu virus known as H5N1 has spread rapidly around the globe, killing countless farmed and wild birds. It has also repeatedly infected wild mammals and been detected in a small number of people. Although the virus remains adapted to birds, scientists worry that it could acquire mutations that help it spread more easily among humans, potentially setting off a pandemic. Many nations have tried to stamp out the virus by increasing biosecurity on farms, quarantining infected premises and culling infected flocks. But the virus has become so widespread in wild birds that it has proved impossible to contain, and some nations have begun vaccinating poultry, although that endeavor presents some logistic and economic challenges. If scientists could engineer resistance into chickens, farmers would not need to routinely vaccinate new batches of birds. Gene editing “promises a new way to make permanent changes in the disease resistance of an animal,” Mike McGrew, an embryologist at the University of Edinburgh’s Roslin Institute and an author of the new study, said at the briefing. “This can be passed down through all the gene-edited animals, to all the offspring.” CRISPR, the gene-editing technology used in the study, is a molecular toolthat allows scientists to make targeted edits in DNA, changing the genetic code at a precise point in the genome. In the new study, the researchers used this approach to tweak a chicken gene that codes for a protein known as ANP32A, which the flu virus hijacks to copy itself. The tweaks were designed to prevent the virus from binding to the protein — and therefore keep it from replicating inside chickens. The edits did not appear to have negative health consequences for the chickens, the researchers said. “We observed that they were healthy, and that the gene-edited hens also laid eggs normally,” said Dr. Alewo Idoko-Akoh, who conducted the research as a postdoctoral researcher at the University of Edinburgh. The researchers then sprayed a dose of flu virus into the nasal cavities of 10 chickens that had not been genetically edited, to serve as the control. (The researchers used a mild version of the virus different from the one that has been causing major outbreaks in recent years.) All of the control chickens were infected with the virus, which they then transmitted to other control chickens they were housed with. When the researchers administered flu virus directly into the nasal cavities of 10 gene-edited chickens, just one of the birds became infected. It had low levels of the virus and did not pass the virus on to other gene-edited birds. “But having seen that, we felt that it would be the responsible thing to be more rigorous, to stress test this and ask, ‘Are these chickens truly resistant?’” Dr. Barclay said. “‘What if they were to somehow encounter a much, much higher dose?’” When the scientists gave the gene-edited chickens a flu dose that was 1,000 times higher, half of the birds became infected. The researchers found, however, that they generally shed lower levels of the virus than control chickens exposed to the same high dose. The researchers then studied samples of the virus from the gene-edited birds that had been infected. These samples had several notable mutations, which appeared to allow the virus to use the edited ANP32A protein to replicate, they found. Some of these mutations also helped the virus replicate better in human cells, although the researchers noted that those mutations in isolation would not be enough to create a virus that was well adapted to humans. Seeing those mutations is “not ideal,” said Richard Webby, who is a bird flu expert at St. Jude Children’s Research Hospital and was not involved in the research. “But when you get to the weeds of these particular changes, then it doesn’t concern me quite so much.” The mutated flu virus was also able to replicate even in the complete absence of the ANP32A protein by using two other proteins in the same family, the researchers found. When they created chicken cells that lacked all three of these proteins, the virus was not able to replicate. Those chicken cells were also resistant to the highly lethal version of H5N1 that has been spreading around the world the last several years. The researchers are now working to create chickens with edits in all three of the genes for the protein family. The big question, Dr. Webby said, was whether chickens with edits in all three genes would still develop normally and grow as fast as poultry producers needed. But the idea of gene editing chickens had enormous promise, he said. “Absolutely, we’re going to get to a point where we can manipulate the host genome to make them less susceptible to flu,” he said. “That’ll be a win for public health.”
Via Juan Lama
|
|
Scooped by
Dr. Stefan Gruenwald
October 9, 2023 8:41 PM
|
Grass may transfer genes from their neighbors in the same way genetically modified crops are made, a new study has revealed. Research, led by the University of Sheffield, is the first to show the frequency at which grasses incorporate DNA from other species into their genomes through a process known as lateral gene transfer. The stolen genetic secrets give them an evolutionary advantage by allowing them to grow faster, bigger or stronger and adapt to new environments quicker. Understanding the rate is important to know the potential impact it can have on a plant's evolution and how it adapts to the environment. Grasses are the most ecologically and economically important group of plants, covering 30% of the earth's terrestrial surface and producing a majority of our food. The Sheffield team sequenced multiple genomes of a species of tropical grass and determined at different time points in its evolution how many genes were acquired -- giving a rate of accumulation. It is now thought these transfers are likely to occur in the same way that some genetically modified crops are made. These findings, published in the journal New Phytologist could inform future work to harness the process to improve crop productivity and make more resilient crops, and have implications on how we view and use controversial GM crops. Dr. Luke Dunning, Research Fellow from the University of Sheffield's School of Biosciences, and senior author of the research, said: "There are many methods to make GM crops, some which require substantial human intervention and some that don't. Some of these methods that require minimal human intervention could occur naturally and facilitate the transfers we have observed in wild grasses. These methods work by contaminating the reproductive process with DNA from a third individual. Our current working hypothesis, and something we plan to test in the near future, is that these same methods are responsible for the gene transfers we document in wild grasses."
|
|
Rescooped by
Dr. Stefan Gruenwald
from Virus World
July 13, 2023 4:07 PM
|
Gene therapies have made spectacular progress in delivering new cures for previously intractable disease, but they remain the world’s most expensive treatments by far. Now companies are replacing the virus in gene therapies with new delivery technologies that promise not only to overcome the limitations of viral vectors but to slash production costs too. Moderna and Generation Bio teamed up recently to develop non-viral gene therapies for liver and immune-related conditions, in a deal that builds on Generation Bio’s lipid nanoparticle (LNP) delivery platform. The collaboration is part of a growing trend to swap virus-driven gene therapeutics for innovative nucleic acid delivery platforms that escape the high costs and technical limitations of viral vectors. Engineered lipid or protein nanobodies, DNA-based nanocarriers, and novel physicochemical methods point the way toward re-dosable genetic medicines (Table 1). Virus-based vectors — particularly those based on adeno-associated virus (AAV) and lentiviruses — have dominated the first wave of gene therapies to gain regulatory approval. But their widespread deployment in dozens of clinical trials has exposed their limitations very clearly. “One of the issues is just the biomass needed to produce a sufficient amount, like AAV at the scale and in the doses needed for large indications,” says Akin Akinc, CEO of Aera Therapeutics, a company developing a protein-nanoparticle-based delivery system. AAV vectors are further limited by their 4.7-kilobase packaging capacity; poor tissue selectivity; risk of liver toxicity; and immunogenicity, which eliminates the possibility of redosing. Lentiviral vectors and, to a lesser extent, AAV vectors also elicit oncogenicity concerns arising from chromosomal integration and insertional mutagenesis. Moreover, the theoretical risk of replication-competent viruses emerging from recombination events during lentiviral production necessitates extensive testing in patients. To provide a viable alternative, non-viral technologies need to satisfy certain well-defined requisites. They must accommodate a large payload and deliver it to specific organs, lack immunogenicity to allow re-dosing, and have a high safety margin and low production costs. Although most are still preclinical, the field is gaining momentum. In recent months, Aera raised $193 million to progress a protein-nanoparticle system based on endogenous human proteins. These assemble spontaneously into capsid-like structures that can deliver a nucleic acid cargo. Intergalactic Therapeutics plans to start clinical trials next year of a gene therapy that employs in vivo electroporation to deliver covalently closed, circular DNA molecules to retinal cells. And ReCode Therapeutics has just entered the clinic with a nebulized LNP that delivers mRNA encoding a protein, DNAI, to the lungs of patients with primary ciliary dyskinesia arising from mutations in dynein axonemal intermediate chain 1 (DNAI1). The global rollout of inexpensive LNP-based mRNA COVID-19 vaccines has demonstrated that it is feasible to manufacture and deliver billions of doses, including repeat doses, of at least one form of nucleic acid. There is, however, a substantial difference between delivering mRNA to antigen-presenting cells locally in muscle and delivering LNP-encapsulated DNA or other cargoes to the cell nucleus within target tissues, be they in the liver, muscle, lungs, heart or other organs, while avoiding triggering innate immune responses. The cell targeting needs to be more sophisticated while the DNA payload may also require modification to ensure it is efficiently localized to the nucleus upon cell entry. For example, Generation Bio’s closed-end DNA (ceDNA) has a covalently closed structure to resist nuclease attack in the cytoplasm and two inverted terminal repeats flanking the transgenes and its regulatory elements to promote nuclear uptake. The liver is another obstacle for any systemically delivered gene therapy not intended for that organ, because it scavenges from the circulation viral and non-viral vectors alike, along with other exogenous and endogenous macromolecules and nanoparticles. The key culprit is the serum protein apolipoprotein E (ApoE), says David Lockhart, president and CSO of ReCode. It promiscuously binds circulating LNPs or other delivery vehicles, and most of them end up in the liver, as it is rich in ApoE-binding low-density lipoprotein receptors. “If you don’t avoid ApoE binding, the liver is a sink,” he says. To direct delivery to different organs, Recode adds a fifth, selective organ targeting (SORT) lipid to the classical four-lipid nanoparticle composition, an approach developed by company co-founder Daniel Siegwart of the University of Texas Southwestern Medical Center. “It’s not one magic lipid — this is a class effect,” Lockhart says. These lipids bind specific serum proteins whose receptors are strongly expressed by the target cells. Thus, adding a lipid that binds the adhesion protein vitronectin improves delivery to the lung, whereas one that binds β2-glycoprotein 1 boosts delivery to the spleen. Generation Bio also adds a fifth, targeting ligand to its LNPs to avoid default trafficking to resident macrophages in the liver and spleen, but the mechanism differs from that of ReCode’s. “It’s a direct receptor engagement,” says Matthew Stanton, CSO at Generation Bio. The company has not yet published details of its ‘stealth’ LNPs but claims it can avoid default trafficking to the liver and spleen, particularly to their resident macrophages. “That’s what got Moderna so excited,” he says. Anjarium Biosciences of Schlieren, Switzerland, achieves cell-specific delivery with antibodies or antibody fragments, which it attaches to its proprietary ‘hybridosme’ nanoparticles. The latter are based on LNPs and exosomes. “We have a composite particle with elements of each,” says CSO and co-founder Joël de Beer. Exosomes have proven to be difficult to load with genetic cargo, he says, but they can be readily modified externally. They are also immunologically silent, given their ubiquity in human circulation and tissues. “The issue with LNPs of course is biodistribution,” he says. But they are efficient carriers of nucleic acids. The company’s aim is to capture their combined benefits while avoiding their shortcomings. Delivery vehicles based on other materials can avoid the issue of liver trafficking from the outset. Code Biotherapeutics is developing a DNA-based nanocarrier that has no inherent liver tropism. Its technology comprises a multivalent structure assembled by hybridizing multiple single strands into double-stranded monomers, which are then cross-linked to form a stable particle. Up to 18 copies of a therapeutic payload and another 18 of a targeting moiety can be attached externally. “We have pretty precise control over how we can build that,” says Brian McVeigh, CEO and co-founder of Code. Yet another option is to build carriers from phage proteins. Gensaic is using three capsid proteins from phage M13, a resident of the human microbiome, in protein-based nanoparticles. “These particles don’t have a pre-existing tropism,” says CEO Lavi Erisson. The protein-based system is readily amenable both to protein engineering methods, for the addition of targeting moieties, and to high-throughput screening methods, for the identification of particles with the desired attributes. The company’s programs remain at an early stage, but efforts in Duchenne muscular dystrophy, cystic fibrosis and central nervous system disorders are underway. Aera’s approach also involves proteins that are present in the body. They are derived from retroelements, such as human endogenous retroviruses and retrotransposons, which constitute a sizeable percentage of the human genome. “By starting with human proteins, we’re starting at a lower immunogenic risk,” says CEO Akinc. Scientific founder Feng Zhang of the Broad Institute of MIT and Harvard has described one such particle, based on a protein called PEG10. Jason Shepherd of the University of Utah has supplied Aera with another protein nanoparticle, based on a protein called Arc, which is expressed in the brain. The company is actively seeking others. “I tend to think there’s going to be better ones out there,” says Akinc. For manufacturing, Aera is employing a cell-free process, unlike the cell-based process that Zhang and colleagues described in their original report. “We can precisely control what we’re putting into the mixture,” says Akinc. The company has not yet disclosed any development programs, but its initial focus will be on short interfering RNA (siRNA), mRNA and gene-editing cargoes. “We are interested in DNA delivery as well,” he says. “I tend to think there’s going to be better ones out there.” Other types of nanoparticles — polymer-based, for instance — do not traffic to the liver by default so developers do not have to deal with that problem. “Typically they’re designed or discovered on a per-tissue basis,” says Sean Kevlahan, CEO and co-founder of Nanite. The rules that determine why a particular particle traffics to a particular tissue are not fully understood, however, and the company is addressing this issue. “If you don’t understand why, then you can’t design them effectively,” he says. To explore the vast chemical space of polymer nanoparticles, the company has developed rapid synthesis and computational methods. It recently secured funding from the Cystic Fibrosis Foundation to identify polymer-based nanoparticles equipped to penetrate the thick mucus that clogs the airways of patients and deliver an mRNA payload. For working ex vivo or for tissues that are readily accessible, scientists may choose to apply physicochemical tools. Apart from one from SQZ Biotech, which is now struggling to stay afloat, most tools remain in preclinical development, although they are edging closer to the clinic. Intergalactic is employing a proprietary needle-like electrode to deliver its C3 double-stranded DNA molecules to target cells. These are assembled in vitro from libraries of transgenes and regulatory elements and then combined into the final product in engineered bacterial cells, which ensures ease of manufacture and low cost of goods. The final DNA product is fully human and does not contain any viral or bacterial sequences. In the company’s lead program, aimed at ABCA4 retinopathy, a solution containing the DNA is administered by subretinal injection and the same area is then exposed to a low-energy electric field. The method is broadly applicable. “Any tissue that can be biopsied is a potential candidate for local [gene] delivery,” says José Lora, Intergalactic’s CSO. Maynooth, Ireland-based Avectas is developing a novel method of ex vivo gene transfer, in which it transiently exposes cells to a solubilizing spray, which contains ethanol and other constituents. This temporarily permeabilizes the cell membrane, allowing macromolecules, such as nucleic acids or gene editing components, to cross into the cytoplasm. Lab tests on T-cells suggest it is on a par with electroporation in terms of gene transfer efficiency, but the transfected cells exhibit less perturbation of gene expression. “Ultimately, that results in a cell that has the potential to be more highly functional,” says Justin McCue, chief technology officer at Avectas. Ultimately, gene therapy’s claims of ‘one and done’ may need revising as transgene expression wanes over time, and if the therapy is virus-based, potential immunogenicity means patients will be unable to receive a second dose. Non-viral gene delivery particles that can reliably demonstrate a lack of immunogenicity could eliminate that issue. What’s more, they could also change the safety equation during clinical development. “It’s not about getting to the maximum-tolerated dose and seeing if you’re efficacious,” says Stanton. Instead, developers can stack multiple “well-tolerated doses” over time, he says, which allows patient-specific dose titration. And if those nanoparticles are also able to evade the liver and reach their target organ, the whole experience of gene therapy will become similar to taking a biologic drug. It will take time and effort to deliver this vision — but it is now starting to take shape. Published in Nat. Biotechnology (June 14, 2023):
Via Juan Lama
|
|
Rescooped by
Dr. Stefan Gruenwald
from Virus World
July 6, 2023 6:16 PM
|
Large DNA viruses of the phylum Nucleocytoviricota infect diverse eukaryotic hosts from protists to humans, with profound consequences for aquatic and terrestrial ecosystems. While nucleocytoviruses are known to be highly diverse in metagenomes, knowledge of their capsid structures is restricted to a few characterized representatives. Here, we visualize giant virus-like particles (VLPs, diameter >0.2 μm) directly from the environment using transmission electron microscopy. We found that Harvard Forest soils contain a higher diversity of giant VLP morphotypes than all hitherto isolated giant viruses combined. These included VLPs with icosahedral capsid symmetry, ovoid shapes similar to pandoraviruses, and bacilliform shapes that may represent novel viruses. We discovered giant icosahedral capsids with structural modifications that had not been described before including tubular appendages, modified vertices, tails, and capsids consisting of multiple layers or internal channels. Many giant VLPs were covered with fibers of varying lengths, thicknesses, densities, and terminal structures. These findings imply that giant viruses employ a much wider array of capsid structures and mechanisms to interact with their host cells than is currently known. We also found diverse tailed bacteriophages and filamentous VLPs, as well as ultra-small cells. Our study offers a first glimpse of the vast diversity of unexplored viral structures in soil and reinforces the potential of transmission electron microscopy for fundamental discoveries in environmental microbiology. Preprint available at bioRxiv (June 30, 2023): https://doi.org/10.1101/2023.06.30.546935
Via Juan Lama
|
|
Scooped by
Dr. Stefan Gruenwald
June 17, 2023 11:17 PM
|
Astronomers have detected, a critical life component, phosphate on Saturn's moon Enceladus, marking a significant step forward in the search for extraterrestrial life. Phosphate is vital for all life on Earth. It forms the backbone of DNA and RNA, the genetic material that carries the instructions for life. It is also a key component of ATP (adenosine triphosphate), the molecule that provides energy for cellular processes, and phospholipids, which form the membranes of cells. The discovery of phosphates on Saturn’s moon Enceladus is the first to report direct evidence of phosphorus on an extraterrestrial ocean world, making it a significant milestone in astrobiology. The detection of phosphates on Saturn’s moon Enceladus was made possible by data from NASA’s Cassini mission. Launched in 1997, Cassini spent 13 years exploring Saturn, its rings, and its moons. One of its most significant discoveries was the detection of a subsurface ocean on Enceladus, which was confirmed by the spacecraft’s observations of geysers erupting from the moon’s south pole. These geysers spew water ice particles into space, some of which supply Saturn’s E ring. The Cassini spacecraft was equipped with the Cosmic Dust Analyzer, which analyzed individual ice grains emitted from Enceladus and sent those measurements back to Earth. In the new study, researchers used data collected from Cassini’s Cosmic Dust Analyzer. They found sodium phosphate molecules in 9 of 345 ice grains Cassini analyzed as it passed through Saturn’s outer E ring. The detection of phosphates on Saturn’s moon Enceladus is a stunning result for astrobiology and a major step forward in the search for life beyond Earth. The high concentration of phosphates in Enceladus’ ocean satisfies one of the strictest requirements in establishing whether celestial bodies are habitable. The discovery of phosphate on Saturn’s moon Enceladus reinforces the idea of the potential habitability of ice-covered ocean worlds across the solar system, beyond surface ocean worlds like Earth. Worlds with an interior ocean like Enceladus can occur over a much wider range of distances, greatly expanding the number of habitable worlds likely to exist across the galaxy. This discovery has profound implications for our understanding of where life might exist beyond Earth. It suggests that the conditions necessary for life as we know it could exist on moons and planets far from the warmth of their parent star, in the cold depths of subsurface oceans. With the discovery of phosphates on Saturn’s moon Enceladus, the next step is clear – we need to go back to Enceladus to see if the habitable ocean is actually inhabited. This finding has made the search for extraterrestrial life in our solar system more exciting and promising. Future missions to Enceladus could involve landers or even submarines designed to probe its subsurface ocean. Such missions could collect and analyze samples directly from the ocean, potentially detecting signs of life such as microorganisms or organic molecules.
|
|
Rescooped by
Dr. Stefan Gruenwald
from Virus World
May 26, 2023 11:59 AM
|
In a recent study posted to the bioRxiv* preprint server, researchers analyzed the viral genomes of 28 endemic viruses to study the evolution of the ability of viruses to evade the neutralizing antibodies elicited by vaccines or previous infections. Background Viruses evolve rapidly and adapt to changing environments due to their high mutation rates and low generation time. Often viruses adapted to different animal hosts infect humans and optimize the methods through which they enter and replicate in the host cell, increasing the human-to-human transmission and evolving into a novel pathogen. The early stages of pandemics are often characterized by high adaptive evolutionary rates, as was seen during the coronavirus disease 2019 (COVID-19) pandemic and outbreaks related to various other respiratory viruses. While some viruses become endemic after adapting to a new host and do not evolve further, other endemic viruses continue to adapt through antigenic evolution, resulting in an arms race between the virus and the human immune system. Since viruses that undergo antigenic evolution pose the risk of repeat infections and increase their ability to evade vaccine-induced immunity, understanding which viruses continue to undergo antigen evolution could help manage future disease outbreaks. About the study The present study used sequence data for each gene in 28 viral genomes to estimate adaptive evolutionary rates. These 28 viruses spanned ten families and were transmitted between humans through various modes. Viruses with potentially high antigenic evolution rates were identified based on the high evolutionary rates for the genes coding for receptor-binding proteins since the receptor-binding region is involved in antibody neutralization and harbors most mutations that allow antigenic escape. The adaptive evolutionary rates were calculated in terms of the number of adaptive mutations in each codon per year, which allowed the adaptive evolutionary rates to be compared across the various genes in the genome and across viruses. The adaptive evolutionary rates of three viruses that were known to undergo antigenic evolution — coronavirus 229E, influenza viruses A/H3N2, and influenza viruses B/Yam — were compared against the evolutionary rates of three antigenically stable viruses, hepatitis A, measles, and influenza C/Yamagata. To understand the patterns of adaptive evolution in recent years, the sequence data for 28 viruses that are pathogenic to humans were obtained and curated. These viruses included deoxyribonucleic acid (DNA) and ribonucleic acid (RNA) viruses and were transmitted between humans through bodily fluids, blood, vectors, fecal-oral, and respiratory routes. The researchers only investigated endemic viruses since they were interested in understanding the antigenic evolution that occurs during the endemic phase and not the initial adaptive phase. The evolutionary rates of the receptor binding protein, which was expected to be highly variable across endemic viruses, and the polymerase gene, which was expected to be conserved, were compared across the 28 viruses. Since the evolution of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has been relatively recent, and the Omicron variant carried a large number of mutations indicating a single fixation event, SARS-CoV-2 was compared with ten other antigenically evolving viruses by comparing the amino-acid substitution rates in the receptor binding protein. Results The results reported that 10 of the 28 viruses undergo adaptive evolution resulting in the antigenic mutations that allow the viruses to escape the immunity induced by previous infections and vaccines. Furthermore, comparing amino-acid substitution rates between SARS-CoV-2 and other viruses revealed that SARS-CoV-2 is evolving and accumulating mutations that cause protein-coding changes at rates much higher than other endemic viruses. Antigenic evolution was found to be more prevalent in RNA viruses. Still, the researchers believe that since the list of viruses included in the study was not comprehensive and comprised only well-studied viruses, determining the proportion of antigenically evolving endemic viruses is difficult. Furthermore, the rate of adaptive mutations might not reflect the phenotypic changes occurring in the viruses, implying that viruses with receptor-binding protein genes that evolve at the same rates might not develop the ability to evade vaccine or infection-induced immunity at the same rates. Conclusions Overall, the findings suggested that many human viruses that have become endemic continue to evolve antigenically, gaining the ability to escape the neutralizing antibodies generated by vaccinations or previous infections. Ten of the 28 viruses investigated in this study showed continued adaptive evolution. In contrast, the amino-acid substitution rates showed that SARS-CoV-2 is evolving faster than the other ten endemic human viruses. Cited ressearch available in bioRxiv (May 22, 2023): https://doi.org/10.1101/2023.05.19.541367
Via Juan Lama
|
|
Scooped by
Dr. Stefan Gruenwald
May 15, 2023 5:35 PM
|
Researchers have developed a biosynthetic “clock” that keeps cells from reaching normal levels of deterioration related to aging. They engineered a gene oscillator that switches between the two normal paths of aging, slowing cell degeneration and setting a record for life extension. Human lifespan is related to the aging of our individual cells. Three years ago a group of University of California San Diego researchers deciphered essential mechanisms behind the aging process. After identifying two distinct directions that cells follow during aging, the researchers genetically manipulated these processes to extend the lifespan of cells. As described April 28, 2023 in Science, they have now extended this research using synthetic biology to engineer a solution that keeps cells from reaching their normal levels of deterioration associated with aging. Cells, including those of yeast, plants, animals and humans, all contain gene regulatory circuits that are responsible for many physiological functions, including aging. “These gene circuits can operate like our home electric circuits that control devices like appliances and automobiles,” said Professor Nan Hao of the School of Biological Sciences’ Department of Molecular Biology, the senior author of the study and co-director of UC San Diego’s Synthetic Biology Institute. However, the UC San Diego group uncovered that, under the control of a central gene regulatory circuit, cells don’t necessarily age the same way. Imagine a car that ages either as the engine deteriorates or as the transmission wears out, but not both at the same time. The UC San Diego team envisioned a “smart aging process” that extends cellular longevity by cycling deterioration from one aging mechanism to another. In this new study, the researchers genetically rewired the circuit that controls cell aging. From its normal role functioning like a toggle switch, they engineered a negative feedback loop to stall the aging process. The rewired circuit operates as a clock-like device, called a gene oscillator, that drives the cell to periodically switch between two detrimental “aged” states, avoiding prolonged commitment to either, and thereby slowing the cell’s degeneration. These advances resulted in a dramatically extended cellular lifespan, setting a new record for life extension through genetic and chemical interventions. As electrical engineers often do, the researchers in this study first used computer simulations of how the core aging circuit operates. This helped them design and test ideas before building or modifying the circuit in the cell. This approach has advantages in saving time and resources to identify effective pro-longevity strategies, compared to more traditional genetic strategies. “This is the first time computationally guided synthetic biology and engineering principles were used to rationally redesign gene circuits and reprogram the aging process to effectively promote longevity,” said Hao. Several years ago the multidisciplinary UC San Diego research team began studying the mechanisms behind cell aging, a complex biological process that underlies human longevity and many diseases. They discovered that cells follow a cascade of molecular changes through their entire lifespan until they eventually degenerate and die. But they noticed that cells of the same genetic material and within the same environment can travel along distinct aging routes. About half of the cells age through a gradual decline in the stability of DNA, where genetic information is stored. The other half ages along a path tied to the decline of mitochondria, the energy production units of cells. The new synthetic biology achievement has the potential to reconfigure scientific approaches to age delay. Distinct from numerous chemical and genetic attempts to force cells into artificial states of “youth,” the new research provides evidence that slowing the ticks of the aging clock is possible by actively preventing cells from committing to a pre-destined path of decline and death, and the clock-like gene oscillators could be a universal system to achieve that.
|
|
Scooped by
Dr. Stefan Gruenwald
May 14, 2023 1:34 PM
|
Scientists have made groundbreaking progress in characterizing the fraction of human DNA that varies between individuals. For more than 20 years, scientists have relied on the human reference genome, a consensus genetic sequence, as a standard against which to compare other genetic data. Used in countless studies, the reference genome has made it possible to identify genes implicated in specific diseases and trace the evolution of human traits, among other things. But it has always been a flawed tool. One of its biggest problems is that about 70 percent of its data came from a single man of predominantly African-European background whose DNA was sequenced during the Human Genome Project, the first effort to capture all of a person's DNA. As a result, it can tell us little about the 0.2 to one percent of genetic sequence that makes each of the seven billion people on this planet different from each other, creating an inherent bias in biomedical data believed to be responsible for some of the health disparities affecting patients today. Many genetic variants found in non-European populations, for instance, aren't represented in the reference genome at all. For years, researchers have called for a resource more inclusive of human diversity with which to diagnose diseases and guide medical treatments. Now scientists with the Human Pangenome Reference Consortium have made groundbreaking progress in characterizing the fraction of human DNA that varies between individuals. As they recently published in Nature, they have assembled genomic sequences of 47 people from around the world into a so-called pangenome in which more than 99 percent of each sequence is rendered with high accuracy. Layered upon each other, these sequences revealed nearly 120 million DNA base pairs that were previously unseen. While it's still a work in progress, the pangenome is public and can be used by scientists around the world as a new standard human genome reference, says The Rockefeller University's Erich D. Jarvis, one of the primary investigators. "This complex genomic collection represents significantly more accurate human genetic diversity than has ever been captured before," he says. "With a greater breadth and depth of genetic data at their disposal, and greater quality of genome assemblies, researchers can refine their understanding of the link between genes and disease traits, and accelerate clinical research." Sourcing diversity Completed in 2003, the first draft of the human genome was relatively imprecise, but it became sharper over the years thanks to filled-in gaps, corrected errors, and advancing sequencing technology. Another milestone was reached last year, when the final eight percent of the genome -- mainly tightly coiled DNA that doesn't code for protein and repetitive DNA regions -- was finally sequenced. Despite this progress, the reference genome remained imperfect, especially with respect to the critical 0.2 to one percent of DNA representing diversity. The Human Pangenome Reference Consortium (HPRC), a government-funded collaboration between more than a dozen research institutions in the United States and Europe, was launched in 2019 to address this problem. At the time, Jarvis, one of the consortium's leaders, was honing advanced sequencing and computational methods through the Vertebrate Genomes Project, which aims to sequence all 70,000 vertebrate species. His and other collaborating labs decided to apply these advances for high-quality diploid genome assemblies to revealing the variation within a single vertebrate: Homo sapiens. To collect a diversity of samples, the researchers turned to the 1000 Genomes Project, a public database of sequenced human genomes that includes more than 2500 individuals representing 26 geographically and ethnically varied populations. Most of the samples come from Africa, home to the planet's largest human diversity. "In many other large human genome diversity projects, the scientists selected mostly European samples," Jarvis says. "We made a purposeful effort to do the opposite. We were trying to counteract the biases of the past." It's likely that gene variants that could inform our knowledge of both common and rare diseases can be found among these populations.
|
|
Scooped by
Dr. Stefan Gruenwald
April 7, 2023 10:55 AM
|
Multicellular organisms typically develop from a single cell into a collection of cells that all have the same genetic material. A research team now discovered a deviation from this developmental hallmark in the yellow crazy ant, Anoplolepis gracilipes. Males are chimeras of haploid cells from two divergent lineages: R and W. R cells are overrepresented in the males’ somatic tissues, whereas W cells are overrepresented in their sperm. Chimerism occurs when parental nuclei bypass syngamy and divide separately within the same egg. When syngamy takes place, the diploid offspring either develops into a queen when the oocyte is fertilized by an R sperm or into a worker when fertilized by a W sperm. This study reveals a mode of reproduction that may be associated with a conflict between lineages to preferentially enter the germ line.
|
|
Rescooped by
Dr. Stefan Gruenwald
from Virus World
March 30, 2023 3:33 PM
|
An Australian company resurrects flesh of lost species to demonstrate potential of meat grown from cells as food source. Recently, they created a mammoth meatball, resurrecting the flesh of the long-extinct animals. The project aims to demonstrate the potential of meat grown from cells, without the slaughter of animals, and to highlight the link between large-scale livestock production and the destruction of wildlife and the climate crisis. The mammoth meatball was produced by Vow, an Australian company, which is taking a different approach to cultured meat. There are scores of companies working on replacements for conventional meat, such as chicken, pork and beef. But Vow is aiming to mix and match cells from unconventional species to create new kinds of meat. The company has already investigated the potential of more than 50 species, including alpaca, buffalo, crocodile, kangaroo, peacocks and different types of fish. The first cultivated meat to be sold to diners will be Japanese quail, which the company expects will be in restaurants in Singapore this year. “We have a behaviour change problem when it comes to meat consumption,” said George Peppou, CEO of Vow . “The goal is to transition a few billion meat eaters away from eating [conventional] animal protein to eating things that can be produced in electrified systems. “And we believe the best way to do that is to invent meat. We look for cells that are easy to grow, really tasty and nutritious, and then mix and match those cells to create really tasty meat.” Tim Noakesmith, who cofounded Vow with Peppou, said: “We chose the woolly mammoth because it’s a symbol of diversity loss and a symbol of climate change.” The creature is thought to have been driven to extinction by hunting by humans and the warming of the world after the last ice age. The initial idea was from Bas Korsten at creative agency Wunderman Thompson: “Our aim is to start a conversation about how we eat, and what the future alternatives can look and taste like. Cultured meat is meat, but not as we know it.” Plant-based alternatives to meat are now common but cultured meat replicates the taste of conventional meat. Cultivated meat – chicken from Good Meat – is currently only sold to consumers in Singapore, but two companies have now passed an approval process in the US. In 2018, another company used DNA from an extinct animal to create gummy bears made from gelatine from a mastodon, another elephant-like animal. Vow worked with Prof Ernst Wolvetang, at the Australian Institute for Bioengineering at the University of Queensland, to create the mammoth muscle protein. His team took the DNA sequence for mammoth myoglobin, a key muscle protein in giving meat its flavor, and filled in the few gaps using elephant DNA. This sequence was placed in myoblast stem cells from a sheep, which replicated to grow to the 20bn cells subsequently used by the company to grow the mammoth meat. “It was ridiculously easy and fast,” said Wolvetang. “We did this in a couple of weeks. Initially, the idea was to produce dodo meat, he said, but the DNA sequences needed do notvyet exist!"
Via Juan Lama
|