

I'm pleased to share with you the first of two publications with Arti Rai on the legal and regulatory landscape around biologics manufacturing patents. Using…
Get Started for FREE
Sign up with Facebook Sign up with X
I don't have a Facebook or a X account



 Your new post is loading...
Your new post is loading... Your new post is loading...
Your new post is loading...
I'm pleased to share with you the first of two publications with Arti Rai on the legal and regulatory landscape around biologics manufacturing patents. Using… 
No comment yet.
Sign up to comment

A preliminary network analysis highlights the complex intellectual property landscape behind mRNA-based COVID-19 vaccines.

Program: Oral and Poster Abstracts Type: Oral Session: 731. Clinical Autologous Transplantation: Building Better Transplant Platforms in Lymphoid Malignancies Hematology Disease Topics & Pathways: multiple myeloma, Biological, Diseases, Therapies, Plasma Cell Disorders, Lymphoid Malignancies, transplantation Monday, December 7, 2020: 10:15 AM Georgia J McCaughan, BMedSc MBBS MMed (Clin Epi)1,2, Steven Tran, BEng MBiomedE3*, Simon Durrant, MBBS FRCP FRCPath4,5,6, Simon J Harrison, MBBS MRCP (UK) PhD FRCPath (UK) FRACP7,8, James Morton, MBBS BSci (Med) FRACP FRCPA GAICD6,9*, Noemi Horvath, MB ChB FRACP FRCPA10, Andrew Spencer11, Ian H. Kerridge, FRACP FRCPA MPhil BA12,13*, Jeremy An Ke Er, MBBS7,14, Luani Barge, MChD, BSc15,16*, Adam Bryant, MBBS (Hon) PhD FRACP FRCPA1,17*, Robin J Filshie, MBChB, PhD, FRACP, FRCPA18, Emily Choong, BSc MBBS19*, Hock Choong Lai, MBBS, FRACP, FRCPA20, Campbell Tiley, MBBS FRACP21, Anthony K Mills, MBBS FRACP FRCPA15,16, Andrew Butler, MB ChB(Edin), MRCP, MRCPath22*, John Moore, MD, FRACP, FRCPA1,23, Mark Hertzberg, MBBS PhD FRACP FRCPA1,24, Glen A Kennedy, MBBS FRACP FRCPA25*, P. Joy Ho, MB.BS. (Hons), D.Phil (Oxon), FRACP, FRCPA, FFSc(RCPA)26, M Hasib Sidiqi, MBBS27, John Bashford, BSc MBBS FRACP FRCPA6,28, David Routledge, BSc (Hons) MBChB MSc PGCert MRCP FRCPath UK RACP7,14, Kerry Taylor, MBBS(Hons) FRACP FRCPA6,9, Cindy H. Lee, MBBS FRACP FRCPA10, Anna Kalff, MBBS29, Wei Xia, MBBS PhD12* and Nada Hamad, MBBS, BSc, MSc, FRACP FRCPA1,23 1Faculty of Medicine, University of New South Wales, Sydney, Australia 2Department of Haematology, St Vincent's Hospital, Waverley, NSW, Australia 3The Australasian Bone Marrow Transplant Recipient Registry, Sydney, NSW, Australia 4Department of Haematology and Bone Marrow Transplantation, Royal Brisbane and Women's Hospital, Brisbane, Australia 5Wesley Clinic Research Centre Stem Cell Transplant Programme, Wesley Hospital, Brisbane, QLD, Australia 6Icon Cancer Care, Brisbane, Australia 7Peter MacCallum Cancer Centre and Royal Melbourne Hospital, Melbourne, Australia 8Melbourne University, Sir Peter MacCallum Dept of Oncology, Melbourne, Australia 9Mater Hospital, Brisbane, Australia 10Department of Haematology, Royal Adelaide Hospital, Adelaide, Australia 11Malignant Haematology and Stem Cell Transplantation Service, Alfred Health, Monash University, Melbourne, Australia 12Department of Haematology, Royal North Shore Hospital, Sydney, Australia 13University of Sydney, Sydney, Australia 14University Hospital Geelong, Geelong, Australia 15Greenslopes Hospital, Brisbane, Australia 16Department of Haematology, Princess Alexandra Hospital, Brisbane, Australia 17Liverpool Hospital, Sydney, Australia 18St. Vincent's Hospital Melbourne, Melbourne, Australia 19Royal Hobart Hospital, Hobart, Australia 20Department of Haematology, Townsville Hospital, Townsville, Australia 21Central Coast Health, Gosford Hospital, Gosford, Australia 22Department of Haematology, Christchurch Hospital, Christchurch, New Zealand 23Department of Haematology, St Vincent's Hospital, Sydney, Australia 24Department of Clinical Haematology, Prince of Wales Hospital, Sydney, Australia 25Department of Haematology, Royal Brisbane and Women's Hospital, Brisbane, Australia 26Sydney Medical School, Royal Prince Alfred Hospital, Camperdown, Australia 27Department of Haematology, Fiona Stanley Hospital, Perth, Australia 28Wesley Hospital, Brisbane, Australia 29Malignant Haematology and Stem Cell Transplantation Service, Alfred Hospital, Prahran, VIC, Australia Background: Upfront autologous stem cell transplantation (ASCT) in multiple myeloma (MM) following induction therapy has been demonstrated to improve progression free survival (PFS) and overall survival (OS). Consideration of transplant eligibility involves assessment of age (typically <70 years), co-morbidities and frailty. In Australia and New Zealand, approximately 70% of all MM patients aged <70 years undergo upfront ASCT compared to approximately 6% aged 70-75 years (Bergin, MRDR Data). We aimed to review the patterns of transplantation in Australia and New Zealand in patients ≥70 years of age and examine survival outcomes and predictors of survival in this cohort. Methods: We analysed 8786 MM patients who received ASCT in Australia and New Zealand between 2001 and 2019. 630 (7.2%) were ≥70 years of age. As there was missing data in the registry, additional data was obtained for 466 ≥70 years of age from 20 sites (performance status (PS), melphalan dose and creatinine clearance (CrCl)). These sites were selected on the basis of number of eligible patients in the registry. Kaplan-Meier analysis was performed to determine PFS and OS. Univariate and multi-variate analysis was performed using Cox proportional hazard model to determine predictors of OS. Results: The baseline patient and disease characteristics are presented in Table 1. The total number of ASCT procedures performed for MM has increased over the study period, and the proportion of ASCT patients ≥70 years has also increased from 5% in 2000-2004 to 11% in 2015-2019 (Figure 1). 33% of patients ≥70 years of age received reduced dose melphalan (140mg/m2 versus 200mg/m2) compared with 10% of patients < 70. Poor PS (ECOG > 1/Karnofsky Performance Score < 80) and CrCl did not significantly predict dose reduction of melphalan. At a median follow-up of 3.8 years, median PFS was 3.3 years (95% CI 2.9-3.8) for those aged ≥70 and 3.4 years (95% CI 3.2-3.6) for those 60-69 (P =0.7). Median OS in those aged ≥70 was 5.6 years (95% CI 4.9-6.3) compared to 6.2 years in those 60-69 (5.8-6.6 years) (P = 0.01). There was no difference in median time to platelet and neutrophil engraftment in patients aged ≥ 70 compared to those < 70. There was no significant difference in transplant related mortality at day 100 in those ≥70 years (1.8%, 95% CI 1-3%) compared to those < 70 (1%, 95% CI 0.7-1.2%) (P = 0.07). OS in all patients aged ≥ 70 (n = 630) was significantly better in patients transplanted between 2010-2019 (n = 451) compared to 2000-2009 (n = 179) (HR 1.62, 1.20-2.19, P = 0.002) (Figure 2) likely correlating with access to bortezomib based induction in 2011/2012 in Australia and New Zealand, and is reflected by an increased proportion of patients achieving a partial response (PR) or better at time of ASCT (Table 1). Increased access to novel agents in the relapsed/refractory MM patients as well as improvements in supportive care also may have contributed. On univariate analysis, other predictors of OS in older patients were poor PS (HR 2.44, 95% CI 1.23-4.81, P = 0.01), higher risk disease (Stage III using Durie-Salmon, ISS or R-ISS) (HR 1.42, 95% CI 1.01-2.00, P < 0.042) and failure to achieve a PR prior to ASCT (HR 1.71, 95% CI 1.01-2.87, P = 0.05). On univariate analysis, melphalan dose did not predict OS (HR 1.35, 95% CI 0.89-2.05, P = 0.2). Multivariate analysis of determinants of OS was performed for the patients in whom we obtained the additional data. Because of missing data for both PS and stage, multivariate analysis incorporating all variables of interest (decade of transplant, melphalan dose, disease status at transplant, CrCl, PS and stage at diagnosis) could only be performed in a subset of patients (n = 163) (Table 2). In this cohort the only significant predictor of OS was poor PS (Table 2). Conclusion: There is increasing utilisation of upfront ASCT in patients aged ≥ 70 in Australia and New Zealand. OS in this group of patients has significantly improved over the study period in keeping with access to bortezomib based induction and novel agents in the relapsed and refractory setting. In a highly selected group of patients ≥70 years of age, ASCT is feasible and associated with excellent PFS and OS. On multivariate analysis, PS was the only predictor of OS. The prospective use of established co-morbidity and frailty scores in assessing transplant eligibility in older patients warrants further evaluation. Disclosures: Harrison: Takeda: Consultancy, Honoraria, Membership on an entity's Board of Directors or advisory committees, Research Funding; Janssen-Cilag: Consultancy, Membership on an entity's Board of Directors or advisory committees, Research Funding; BMS: Consultancy, Honoraria; Haemalogix: Consultancy; AbbVie: Consultancy, Honoraria, Membership on an entity's Board of Directors or advisory committees, Research Funding; GSK: Consultancy, Honoraria, Membership on an entity's Board of Directors or advisory committees, Research Funding; CRISPR Therapeutics: Consultancy, Honoraria; F. Hoffmann-La Roche: Consultancy, Honoraria; Janssen: Honoraria; Novartis: Consultancy, Honoraria, Patents & Royalties: wrt panobinostat; Amgen: Consultancy, Honoraria, Membership on an entity's Board of Directors or advisory committees, Research Funding; Celgene: Consultancy, Honoraria, Membership on an entity's Board of Directors or advisory committees, Research Funding. Spencer: AbbVie, Amgen, Celgene, Haemalogix, Janssen, Sanofi, SecuraBio, Specialised Therapeutics Australia, Servier and Takeda: Honoraria; Celgene, Janssen and Takeda: Speakers Bureau; AbbVie, Celgene, Haemalogix, Janssen, Sanofi, SecuraBio, Specialised Therapeutics Australia, Servier and Takeda: Consultancy; Amgen, Celgene, Haemalogix, Janssen, Servier and Takeda: Research Funding. Mills: Celgene: Honoraria; Novartis: Honoraria, Other: Meeting sponsorship; AstraZeneca: Honoraria; Abbvie: Membership on an entity's Board of Directors or advisory committees. Hertzberg: Takeda: Honoraria, Membership on an entity's Board of Directors or advisory committees; Roche: Honoraria, Membership on an entity's Board of Directors or advisory committees, Other: Support of parent study and funding of editorial support; MSD: Membership on an entity's Board of Directors or advisory committees; Gilead: Membership on an entity's Board of Directors or advisory committees; BMS: Honoraria; Abbvie: Honoraria; Janssen: Honoraria, Membership on an entity's Board of Directors or advisory committees. Sidiqi: Amgen: Honoraria; Janssen: Honoraria; Celgene: Honoraria, Other: Travel grant. Kalff: Celgene: Honoraria; Janssen: Honoraria; Amgen: Honoraria; CSL: Honoraria; Roche: Honoraria. Hamad: Novartis: Honoraria; Abbvie: Honoraria. See more of: 731. Clinical Autologous Transplantation: Building Better Transplant Platforms in Lymphoid Malignancies See more of: Oral and Poster Abstracts << Previous Abstract | Next Abstract *signifies non-member of ASH

Heart disease is the primary cause of death worldwide, principally because the heart has minimal ability to regenerate muscle tissue. Myocardial infarction (heart attack) caused by coronary artery disease leads to heart muscle loss and replacement with scar tissue, and the heart's pumping ability is permanently reduced. Breakthroughs in stem cell biology in the 1990s and 2000s led to the hypothesis that heart muscle cells (cardiomyocytes) could be regenerated by transplanting stem cells or their derivatives. It has been ∼18 years since the first clinical trials of stem cell therapy for heart repair were initiated (1), mostly using adult cells. Although cell therapy is feasible and largely safe, randomized, controlled trials in patients show little consistent benefit from any of the treatments with adult-derived cells (2). In the meantime, pluripotent stem cells have produced bona fide heart muscle regeneration in animal studies and are emerging as leading candidates for human heart regeneration. In retrospect, the lack of efficacy in these adult cell trials might have been predicted. The most common cell type delivered has been bone marrow mononuclear cells, but other transplanted cell types include bone marrow mesenchymal stromal cells and skeletal muscle myoblasts, and a few studies have used putative progenitors isolated from the adult heart itself. Although each of these adult cell types was originally postulated to differentiate directly into cardiomyocytes, none of them actually do. Indeed, with the exception of skeletal muscle myoblasts, none of these cell types survive more than a few days in the injured heart (see the figure). Unfortunately, the studies using bone marrow and adult resident cardiac progenitor cells were based on a large body of fraudulent work (3), which has led to the retraction of >30 publications. This has left clinical investigators wondering whether their trials should continue, given the lack of scientific foundation and the low but measurable risk of bleeding, stroke, and infection. Additionally, investigators have struggled to explain the beneficial effects of adult cell therapy in preclinical animal models. Because none of these injected cell types survive and engraft in meaningful numbers or directly generate new myocardium, the mechanism has always been somewhat mysterious. Most research has focused on paracrine-mediated activation of endogenous repair mechanisms or preventing additional death of cardiomyocytes. Multiple protein factors, exosomes (small extracellular vesicles), and microRNAs have been proposed as the paracrine effectors, and an acute immunomodulatory effect has recently been suggested to underlie the benefits of adult cell therapy (4). Regardless, if cell engraftment or survival is not required, the durability of the therapy and need for actual cells versus their paracrine effectors is unclear. Of particular importance to clinical translation is whether cell therapy is additive to optimal medical therapy. This remains unclear because almost all preclinical studies do not use standard medical treatment for myocardial infarction. Given the uncertainties about efficacy and concerns over the veracity of much of the underlying data, whether agencies should continue funding clinical trials using adult cells to treat heart disease should be assessed. Perhaps it is time for proponents of adult cardiac cell therapy to reconsider the approach. Pluripotent stem cells (PSCs) include embryonic stem cells (ESCs) and their reprogrammed cousins, induced pluripotent stem cells (iPSCs). In contrast to adult cells, PSCs can divide indefinitely and differentiate into virtually every cell type in the human body, including cardiomyocytes. These remarkable attributes also make ESCs and iPSCs more challenging to control. Through painstaking development, cell expansion and differentiation protocols have advanced such that batches of 1 billion to 10 billion pharmaceutical-grade cardiomyocytes, at >90% purity, can be generated. Preclinical studies indicate that PSC-cardiomyocytes can remuscularize infarcted regions of the heart (see the figure). The new myocardium persists for at least 3 months (the longest time studied), and physiological studies indicate that it beats in synchrony with host myocardium. The new myocardium results in substantial improvement in cardiac function in multiple animal models, including nonhuman primates (5). Although the mechanism of action is still under study, there is evidence that these cells directly support the heart's pumping function, in addition to providing paracrine factors. These findings are in line with the original hope for stem cell therapy—to regenerate lost tissue and restore organ function. Additional effects, such as mechanically buttressing the injured heart wall, may also contribute. Breakthroughs in cancer immunotherapy have led to the adoption of cell therapies using patient-derived (autologous) T cells that are genetically modified to express chimeric antigen receptors (CARs) that recognize cancer cell antigens. CAR T cells are the first U.S. Food and Drug Administration (FDA)–approved, gene-modified cellular pharmaceutical (6). The clinical and commercial success of autologous CAR T cell transplant to treat B cell malignancies has opened doors for other complex cell therapies, including PSC derivatives. There is now a regulatory path to the clinic, private-sector funding is attracted to this field, and clinical investigators in other areas are encouraged to embrace this technology. Indeed, the first transplants of human ESC-derived cardiac progenitors, surgically delivered as a patch onto the heart's surface, have been carried out (7). In the coming years, multiple attempts to use PSC-derived cardiomyocytes to repair the human heart are likely. What might the first human trials look like? These studies will probably employ an allogeneic (non-self), off-the-shelf, cryopreserved cell product. Although the discovery of iPSCs raised hopes for widespread use of autologous stem cell therapies, the current technology and regulatory requirements likely make this approach too costly for something as common as heart disease, although this could change as technology and regulations evolve. Given that it would take at least 6 months to generate a therapeutic dose of iPSC-derived cardiomyocytes, such cells could only be applied to patients whose infarcts are in the chronic phase where scarring (fibrosis) and ventricular remodeling are complete. Preclinical data indicate that chronic infarcts benefit less from cardiomyocyte transplantation than do those with active wound-healing processes. The need for allogeneic cells raises the question of how to prevent immune rejection, both from innate immune responses in the acute phase of transplantation or from adaptive immune responses that develop more slowly through the detection of non-self antigens presented by major histocompatibility complexes (MHCs). A current strategy is the collection of iPSCs from patients who have homozygous MHC loci, which results in exponentially more MHC matches with the general population. However, studies in macaque monkeys suggest that MHC matching will be insufficient. In a macaque model of brain injury, immunosuppression was required to prevent rejection of MHC-matched iPSC-derived neurons (8). Similarly, MHC matching reduced the immunogenicity of iPSC-derived cardiomyocytes transplanted subcutaneously or into the hearts of rhesus macaques, but immunosuppressive drugs were still required to prevent rejection (9). Numerous immune gene editing approaches have been proposed to circumvent rejection, including preventing MHC class I and II molecule expression, overexpressing immunomodulatory cell-surface factors, such CD47 and human leukocyte antigen E (HLA-E) and HLA-G (two human MHC molecules that promote maternal-fetal immune tolerance), or engineering cells to produce immunosuppressants such as programmed cell death ligand 1 (PDL1) and cytotoxic T lymphocyte–associated antigen 4 (CTLA4) (10). These approaches singly or in combination seem to reduce adaptive immune responses in vitro and in mouse models. Overexpressing HLA-G or CD47 also blunts the innate natural killer cell–mediated response that results from deleting MHC class I genes (11). However, these manipulations are not without theoretical risks. It could be difficult to clear viral infections from an immunostealthy “patch” of tissue, and possible tumors resulting from engraftment of PSCs might be difficult to clear immunologically. Ventricular arrhythmias have emerged as the major toxicity of cardiomyocyte cell therapy. Initial studies in small animals showed no arrhythmic complications (probably because their heart rates are too fast), but in large animals with human-like heart rates, arrhythmias were consistently observed (5, 12). Stereotypically, these arrhythmias arise a few days after transplantation, peak within a few weeks, and subside after 4 to 6 weeks. The arrhythmias were well tolerated in macaques (5) but were lethal in a subset of pigs (12). Electrophysiological studies indicate that these arrhythmias originate in graft regions from a source that behaves like an ectopic pacemaker. Understanding the mechanism of these arrhythmias and developing solutions are major areas of research. There is particular interest in the hypothesis that the immaturity of PSC-cardiomyocytes contributes to these arrhythmias, and that their maturation in situ caused arrhythmias to subside. A successful therapy for heart regeneration also requires understanding the host side of the equation. PSC-derived cardiomyocytes engraft despite transplantation into injured myocardium that is ischemic with poor blood flow. Although vessels eventually grow in from the host tissue, normal perfusion is not restored. Achieving a robust arterial input will be key to restoring function, which may require cotransplanting other cell populations or tissue engineering approaches (13, 14). Most PSC-mediated cardiac cell therapy studies have been performed in the subacute window, equivalent to 2 to 4 weeks after myocardial infarction in humans. At this point, there has been insufficient time for a substantial fibrotic response. Fibrosis has multiple deleterious features, including mechanically stiffening the tissue and creating zones of electrical insulation that can cause arrhythmias. Extending this therapy to other clinical situations, such as chronic heart failure, will require additional approaches that address the preexisting fibrosis. Cell therapy may again provide an answer because CAR T cells targeted to cardiac fibroblasts reduced fibrosis (15). Developing a human cardiomyocyte therapy for heart regeneration will push the limits of cell manufacturing. Each patient will likely require a dose of 1 billion to 10 billion cells. Given the widespread nature of ischemic heart disease, 105 to 106 patients a year are likely to need treatment, which translates to 1014 to 1016 cardiomyocytes per year. Growing cells at this scale will require introduction of next generation bioreactors, development of lower-cost media, construction of large-scale cryopreservation and banking systems, and establishment of a robust supply chain compatible with clinical-grade manufacturing practices. Beyond PSC-cardiomyocytes, other promising approaches include reactivating cardiomyocyte division and reprogramming fibroblasts to form new cardiomyocytes. However, these approaches are at an earlier stage of development, and currently, PSC-derived cardiomyocyte therapy is the only approach that results in large and lasting new muscle grafts. The hurdles to this treatment are known, and likely addressable, thus multiple clinical trials are anticipated. http://www.sciencemag.org/about/science-licenses-journal-article-reuse This is an article distributed under the terms of the Science Journals Default License. References and Notes ↵ P. Menasché, Nat. Rev. Cardiol. 15, 659 (2018).OpenUrlCrossRef ↵ K. Nakamura, C. E. Murry, Circ. J. 83, 2399 (2019).OpenUrl ↵ K. R. Chien et al., Nat. Biotechnol. 37, 232 (2019).OpenUrl ↵ R. J. Vagnozzi et al., Nature 577, 405 (2020).OpenUrl ↵ Y. W. Liu et al., Nat. Biotechnol. 36, 597 (2018).OpenUrlCrossRefPubMed ↵ M. M. Boyiadzis et al., J. Immunother. Cancer 6, 137 (2018). ↵ P. Menasché et al., Eur. Heart J. 36, 2011 (2015).OpenUrlCrossRefPubMed ↵ R. Aron Badin et al., Nat. Commun. 10, 4357 (2019).OpenUrl ↵ T. Kawamura et al., Stem Cell Reports 6, 312 (2016).OpenUrl ↵ R. Lanza et al., Nat. Rev. Immunol. 19, 723 (2019).OpenUrl ↵ T. Deuse et al., Nat. Biotechnol. 37, 252 (2019).OpenUrl ↵ R. Romagnuolo et al., Stem Cell Reports 12, 967 (2019).OpenUrl ↵ J. Bargehr et al., Nat. Biotechnol. 37, 895 (2019).OpenUrlCrossRef ↵ M. A. Redd et al., Nat. Commun. 10, 584 (2019).OpenUrlCrossRef ↵ H. Aghajanian et al., Nature 573, 430 (2019).OpenUrlCrossRef Acknowledgments: C.E.M. and W.R.M. are scientific founders of and equity holders in Sana Biotechnology. C.E.M. is an employee of Sana Biotechnology. W.R.M. is a consultant for Sana Biotechnology. C.E.M. and W.R.M. hold issued and pending patents in the field of stem cell and regenerative biology.

The purpose of this study is to mine CAR-T patents and therapies under development, to design a landscape of the sector and to understand key therapy segments a...
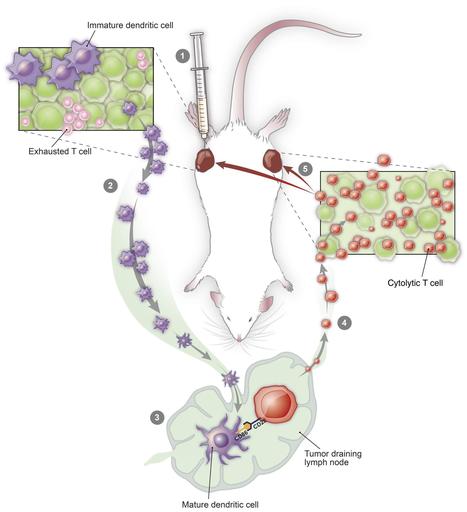
From
www

Research ArticleOncology Free access | 10.1172/JCI128562 In situ vaccination with defined factors overcomes T cell exhaustion in distant tumors Danny N. Khalil,1,2,3,4 Nathan Suek,1 Luis Felipe Campesato,1 Sadna Budhu,1 David Redmond,1 Robert M. Samstein,5 Chirag Krishna,6 Katherine S. Panageas,7 Marinela Capanu,7 Sean Houghton,1 Daniel Hirschhorn,1 Roberta Zappasodi,1,2 Rachel Giese,1,8 Billel Gasmi,9 Michael Schneider,1 Aditi Gupta,1 James J. Harding,3,4 John Alec Moral,8 Vinod P. Balachandran,2,10 Jedd D. Wolchok,1,2,3,4 and Taha Merghoub1,2,3,4 First published July 22, 2019 - More info Abstract Irreversible T cell exhaustion limits the efficacy of programmed cell death 1 (PD-1) blockade. We observed that dual CD40-TLR4 stimulation within a single tumor restored PD-1 sensitivity and that this regimen triggered a systemic tumor-specific CD8+ T cell response. This approach effectively treated established tumors in diverse syngeneic cancer models, and the systemic effect was dependent on the injected tumor, indicating that treated tumors were converted into necessary components of this therapy. Strikingly, this approach was associated with the absence of exhausted PD-1hi T cells in treated and distant tumors, while sparing the intervening draining lymph node and spleen. Furthermore, patients with transcription changes like those induced by this therapy experienced improved progression-free survival with anti–PD-1 treatment. Dual CD40-TLR4 activation within a single tumor is thus an approach for overcoming resistance to PD-1 blockade that is unique in its ability to cause the loss of exhausted T cells within tumors while sparing nonmalignant tissues. Graphical Abstract Introduction Immune checkpoint blockade (ICB) has improved outcomes for patients with diverse cancers, yet most patients with common cancers still do not show a clinical response. Features of malignant cells (1) as well as the immune infiltrate (2) play an important role in mediating resistance. In the latter category, T cell exhaustion acts as a key barrier to effective immunotherapy (3). Here, we propose that antigen-presenting cell (APC) activation status within a single tumor can impact tumor-associated T cell exhaustion systemically. We sought to overcome the hurdle imposed by T cell exhaustion through local APC activation using low-dose intratumoral therapy. We hypothesized that such an approach would augment the activity of programmed cell death 1 (PD-1) blockade for 2 reasons. First, because activating APCs exposed to tumor antigens may prime new waves of tumor-specific T cells; and second, because PD-1 has been shown to regulate CD28 signaling in T cells (4, 5), which is triggered by the costimulatory ligands CD80 and CD86 on activated APCs (6). Using syngeneic murine models, we identified an intratumoral treatment approach using dual CD40-TLR4 stimulation that can overcome tumor-associated T cell exhaustion. Remarkably, this approach eliminated phenotypically exhausted CD8+ T cells at both treated and distant (so-called abscopal; refs. 7, 8) tumors, while sparing lymphoid organs such as the intervening draining lymph node (DLN) and spleen. Results Defined factors control local and distant tumors. To investigate the possibility of a causal relationship between APC activation, particularly CD86 expression, and an antitumor immune response, we used monophosphoryl lipid A (MPL), a low-toxicity TLR4 agonist that induces APC activation in human and murine APCs (9, 10). We used C57BL/6 bone marrow–derived APCs to determine whether MPL induces CD86 expression and phagocytosis in vitro in the CD11chi cell population (Figure 1A). We next examined whether these effects were maintained in tumors in vivo using the syngeneic B16F10 melanoma model. We found that MPL retains the ability to augment phagocytosis, as it induced the uptake of intratumorally implanted latex particles (11) by endogenous CD11chi phagocytic cells (Figure 1B). However, MPL did not retain the ability to activate APCs in the tumor microenvironment (Figure 1B). Given the potent role that CD40 can play in APC activation (12) and its ability to sensitize DCs to TLR stimulation (13), we tested the effect of an agonistic CD40 mAb on APC activation in vivo when administered intratumorally. We found that anti-CD40 induced the activation of CD11chi cells following intratumoral administration (Figure 1B) in this system. Figure 1 Agents that promote phagocytosis and APC activation, but not direct tumor cell lysis, control local and distant tumors. (A) Bone marrow–derived myeloid cells were treated with MPL or vehicle for 16 hours and incubated with FITC-labeled latex beads. Flow cytometry was performed to determine the fraction of CD11chi cells that phagocytosed FITC-labeled beads and the median fluorescence intensity (MFI) of CD86 expression (n = 3 – 5/group). (B) C57BL/6 mice were implanted intradermally with 5 × 105 B16F10 cells. On day 8, FITC-labeled latex beads were coinjected intratumorally with vehicle, MPL, or anti-CD40. Twenty-four hours later, the CD11chi cell population was analyzed in tumors (left) for phagocytosis (n = 5/group) and in DLNs (right) for CD86 expression (n = 4/group). (C) Treatment schedule: intratumoral biweekly treatments, with or without intraperitoneal anti–PD-1, were started once bilateral tumors were established; treatment was continued for 4 weeks. (D) Individual growth curves of treated and distant tumors in animals treated with MPL and anti-CD40 (n = 10/group). (E) Average tumor growth curves comparing MPL and anti-CD40 with constituent monotherapies (n = 10/group). (F) Viability of B16F10 cells treated in vitro with MPL, anti-CD40, or gemcitabine for 72 hours. (G) Growth of treated and distant tumors upon addition of anti–PD-1 (n = 10/group). *P ≤ 0.0, **P ≤ 0.01, ***P ≤ 0.001, and ****P ≤ 0.0001, by unpaired, 2-tailed Student’s t test. Hypothesizing that intratumoral administration of agents mediating phagocytosis and APC activation would trigger a systemic antitumor immune response, we used a bilateral tumor approach (Figure 1C). This allowed us to distinguish the impact of therapy on the treated tumor from that on the distant tumor in animals bearing established, concurrently implanted tumors. We found that the combination of MPL and anti-CD40 eradicated or delayed the growth of treated and distant tumors, respectively (Figure 1D), and that this combination conferred greater antitumor activity than did either anti-CD40 or MPL monotherapy at both the treated and distant tumors (Figure 1E). To assess the possibility that MPL or anti-CD40 was directly cytotoxic, we asked whether these agents directly affect B16F10 viability in vitro. In contrast to oncolytic agents used for in situ vaccination (14), we found that neither MPL nor anti-CD40 demonstrated direct cytolytic activity (Figure 1F). Given the potential of activated APCs to prime antitumor T cells, we next asked whether the addition of anti–PD-1 treatment would augment treatment efficacy in this PD-1–resistant (15, 16) model. We found that addition of anti–PD-1 improved tumor control at both the treated and distant tumors (Figure 1G). Treatment efficacy depends on BATF3+ DCs and CD8+ T cells. To understand the mechanism through which the anti-CD40, MPL, and anti–PD-1 (CMP) regimen mediates antitumor activity, we analyzed distant tumors after 1 week of treatment and found that CMP-treated, but not isotype-treated, animals developed lymphocytic infiltrates deep within the tumor (Figure 2A). This corresponded with an increased fraction of CD8+ T cells and greater proliferation within this population (Figure 2A). The distant tumors continued to be enriched for CD8+ T cells after 3 weeks of treatment, and this enrichment became more pronounced by 6 weeks (Figure 2B), at which point only the treated animals were alive. Figure 2 CMP combination therapy augments APC activation and nodal accumulation followed by a systemic CD8+ T cell response. (A) Using the bilateral tumor model, distant tumors from isotype- (Control) and CMP-treated (Trx) animals were assessed by H&E staining after 1 week of treatment (scale bars: 50 μm) and by flow cytometry to quantify CD8+ T cell infiltrates and the fraction of this cell population expressing Ki67 (n = 4/group). (B) Distant tumors were analyzed by immunofluorescence (IF) at 3 and 6 weeks for CD4 (green), FoxP3 (yellow), and CD8 (red) cell populations (scale bars: 50 μm). Quantification of the CD8+ fraction of DAPI+ cells in IF images (n = 3–10/group). N/A, no remaining live animals. (C) Growth of treated and distant tumors from WT or Rag1–/– C57BL/6 animals (n = 10/group). (D) Growth of treated and distant tumors from mice depleted of CD4+ and CD8+ T cells. Peripheral blood was collected to confirm the absence of corresponding cell populations (n = 10/group). (E) Tumor growth in mice bearing treated WT B16F10 and distant B78H10 tumors (n = 9–10/group). (F) Mice previously cured of unilateral B16F10 tumors with CMP treatment and age-matched naive controls were implanted with tumors on day 90 (n = 8–10/group). Adjacent panel shows fur depigmentation at the site of the initial cured tumor (green arrowhead) and at the site of post-treatment tumor reimplantation (red arrowhead). (G) CD86 expression in the CD11chi cell population in the treated tumor and DLN (n = 4/group). (H) Fraction of CD11chi cells among live CD45+ cells in the treated tumor and DLN (n = 4/group). (I) Tumor growth of WT or Batf3–/– C57BL/6 animals bearing B16F10 tumors treated with isotype or CMP (n = 10/group). *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, and ****P ≤ 0.0001, by unpaired, 2-tailed Student’s t test. Given the evidence of a proliferative CD8+ T cell infiltrates within abscopal tumors, we next asked whether this cell population was necessary for treatment efficacy. We found that CMP efficacy was lost in Rag1–/– mice lacking mature lymphocytes (Figure 2C) and mice depleted of CD8+, but not CD4+, T cells (Figure 2D). Given the necessity of CD8+ T cells in mediating treatment efficacy, we next used the MHC-deficient B16 cell line B78H1 (17) to confirm MHC-dependent killing of abscopal tumor cells. Mice were implanted with B16 at the treated site and B78H1 at the distant site. While local B16 tumors regressed, we observed that treatment efficacy was indeed lost at the distant B78H1 tumors (Figure 2E and Supplemental Figure 1; supplemental material available online with this article; https://doi.org/10.1172/JCI128562DS1). To assess for persistent immunity, the cured animals treated initially with CMP were reimplanted with B16F10 in the contralateral flank 90 days after treatment. All such animals fully resisted tumor reimplantation, whereas all of the naive animals developed growing tumors. Interestingly, autoimmune fur depigmentation occurred at both the site of the initial tumor and the site of tumor reimplantation, despite the absence of ongoing therapy, in a subset of animals (Figure 2F). We next examined the effect of CMP treatment on APCs to identify a cell population that may be interacting with the CD8+ T cells controlling tumor growth. We found that the CD11chi immune cell population, an important category of APCs within the tumor microenvironment (18), became activated within 3 hours of treatment and that activation persisted for 24 hours within the tumor (Figure 2G). As this occurred, we observed a delayed wave of CD11chi activation in the DLN that was evident 24 and 48 hours after treatment (Figure 2G). Strikingly, during these waves of activation, the CD11chi cell population was reduced within the tumor and concurrently accumulated within the DLN (Figure 2H). This pattern of CD11chi cell activation in the tumor followed by its activation in the DLN, together with the depletion of this cell population in the tumor and concurrent accumulation in the DLN, is consistent with nodal trafficking of activated APCs (19). Given these findings in the CD11chi cell population and the necessity of CD8+ T cells for tumor control, we asked whether BATF3+ DCs were necessary for treatment efficacy, as these DCs efficiently cross-present antigens to CD8+ T cells (20–22). Using animals deficient for BATF3+ DCs (20), we found that treatment efficacy was indeed lost in this system (Figure 2I), confirming the necessity of this DC population for treatment efficacy. CMP acts on tumor-specific T cells. We next sought to determine whether CMP induced a tumor-specific T cell response, as would be expected if BATF3+ DCs exposed to tumor antigens were priming naive CD8+ T cells. We performed an IFN-γ enzyme-linked immune absorbent spot (ELISpot) assay on CD8+ T cells sorted from treated animals 1 week after treatment and found that CMP selectively increased the response to B16F10 relative to syngeneic control tumor cells (Figure 3A). Next, we adoptively transferred Pmel-1 CD8+ T cells, specific to the melanoma differentiation antigen gp100, derived from T cell receptor–transgenic (TCR-transgenic) mice (23) into B16F10 tumor–bearing animals. As expected, we found that treatment induced proliferation in this B16F10-targeting T cell population (Figure 3B). To further assess the functional impact of CMP treatment, we used an ex vivo killing assay (24), in which CD8+ T cell cells were isolated from isotype- or CMP-treated animals. Since T cell quantities were held constant ex vivo, we were able to determine whether the cytolytic capacity of individual T cells was augmented by treatment and found that CMP enhanced the ability of individual T cells to lyse B16F10 tumor cells (Figure 3C). To determine whether CMP induces the selective expansion of endogenous T cells specific to the tumor and assess the contribution of individual treatment components, we used combinatorial encoding of MHC multimers (25). This system, capable of detecting rare endogenous T cells specific to an antigen of interest, revealed that CMP markedly expanded endogenous gp100-specific T cells that were nearly undetectable at baseline (Figure 3D). Figure 3 CMP stimulates tumor-specific CD8+ T cells. (A) B16F10 tumor–bearing mice were treated for 1 week with CMP or isotype control. CD8+ T cells were purified from spleens and restimulated with irradiated ID8 or B16F10 stimulator cells. Shown are IFN-γ+ cytotoxic T lymphocytes (CTLs) in CMP and isotype samples for both tumor cell types (n = 3–5/group). (B) One week after tumor challenge, mice bearing B16F10 tumors were adoptively transferred with 2 × 106 CellTrace Violet–labeled (CTV-labeled) Pmel-1 CD8+ T cells purified from naive TCR-transgenic mice. A single treatment with CMP or isotype was administered on day 8, and LNs were harvested on day 11 to assess proliferation. Quantification of undiluted cells is shown in the CTV dilution histograms (n = 4–5/group), which are representative of 2 experiments. (C) CD8+ T cells were purified from distant tumors of animals treated for 1 week with CMP and used for an ex vivo collagen-fibrin gel–based killing assay. CD8+ T cells were coincubated with B16 tumor cells, and tumor cell numbers were assessed using a clonogenic assay to determine the proportion of B16 cells killed and the killing constant. (D) Tumor-bearing mice were sacrificed after 1 week of CMP treatment, and DLNs were stained with H2-Db MHC class I multimers bearing peptides from the melanoma differentiation antigen gp100. Quantification of endogenous gp100-specific CD8+ T cells relative to the total CD8+ T cell population is shown in representative plots (n = 3–5/group). **P ≤ 0.01 and ****P ≤ 0.0001, by unpaired, 2-tailed Student’s t test. Having determined that CMP therapy augments the tumor-specific T cell response in a B16F10 model, we next asked whether this therapy is effective in other syngeneic models and found that it was. We observed activity in bilateral syngeneic models of colorectal, hepatocellular, and bladder cancer (Figure 4A) applying the same bilateral tumor approach used for the B16F10 tumors (Figure 1C). We next used the orthotopic, syngeneic KPC pancreatic cancer model, in which anti-CD40 monotherapy has had little to no efficacy (26). In animals bearing established KPC tumors, we found that intravenous administration was effective (without a dose increase compared with intratumoral treatment) and even induced cures in nearly half of the treated mice, whereas PD-1 monotherapy was not significantly more effective than isotype control antibody treatment (Figure 4B). Figure 4 CMP is active across tumor models but inactive in unmatched distant tumors. (A) Average growth curves of animals bearing bilateral MC38, MB49, and Hep-55.1c tumors (n = 10/group). (B) Kaplan-Meier curves show survival rates of C57BL/6 animals bearing established orthotopic KPC pancreatic tumors treated intravenously with anti-CD40 and MPL (n = 10/group). (C) Bilateral tumor model with distant B16F10 tumors and either MC38, Hep55.1c, or B16F10 (positive control) as the treated tumor (n = 10/group). *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, and ****P ≤ 0.0001, by unpaired, 2-tailed Student’s t test. Treatment efficacy is lost in unmatched bilateral syngeneic tumors. Having seen efficacy in diverse tumor models, we returned to the bilateral system, but now with unmatched syngeneic tumors at each flank (Figure 4C). Importantly, since we previously found that distant B16F10 tumors are sensitive to CMP treatment when the treated tumor is also B16F10, we used distant B16F10 tumors for all unmatched bilateral tumor experiments. This would allow us to exclude the possibility that the injected agents control distant tumors by simple diffusion and direct contact. The syngeneic tumor cell lines Hep-55.1C and MC38 (hepatocellular and colorectal carcinoma lines, respectively) were used as the treated tumors and were implanted concurrently with the distant B16F10 tumors. Consistent with tumor-specific T cell–mediated killing, these experiments revealed that when different syngeneic tumors were used as the treated tumor, distant B16F10 tumors were not controlled with CMP relative to PD-1 blockade alone (Figure 4C), indicating that the treated tumor itself is a necessary component of this therapy. CMP is associated with the absence of exhausted T cells selectively within tumors. To further characterize the systemic impact of treatment, we conducted RNA profiling of distant tumors 1 week after starting CMP treatment. We observed transcriptional changes consistent with robust T cell activity (Figure 5A). For example, granzyme K, a gene expressed in activated T cells and associated with cytolytic activity (27, 28), was the most significantly upregulated gene in distant tumors with CMP treatment versus isotype, whereas it was not significantly upregulated with anti–PD-1 alone (Figure 5A). To better understand these results, we asked whether transcriptional changes in the distant tumor at 1 week were associated with a particular T cell phenotype. Using annotated gene sets from the Molecular Signatures Database (29) defined by genes upregulated in effector versus exhausted CD8+ T cells during chronic infection (29), we found clear separation among treatment samples along the exhausted to effector spectrum. Encouragingly, samples from animals treated with PD-1 monotherapy (which is known to reinvigorate a subset of exhausted T cells) (30) appeared less exhausted than those treated with isotype control and segregated away from these samples with unsupervised clustering (Figure 5B). Strikingly, all 4 samples treated with CMP were shifted further away from an exhausted expression pattern than those treated with anti–PD-1, despite having administered the intratumoral therapy into the contralateral tumor (Figure 5B). Figure 5 CMP selectively eliminates PD-1hi T cells in tumors while sparing nontumor tissues. (A) Expression profiling of distant tumors in bilateral tumor–bearing mice after 1 week of CMP treatment (n = 4/group). Shown are transcriptional changes induced by PD-1 monotherapy or CMP therapy relative to isotype control. (B) Heatmap generated using unsupervised clustering based on annotated gene sets from the Molecular Signatures Database defined by genes upregulated in effector versus exhausted CD8+ T cells during chronic infection (FDR q value = 7.35 × 10–11). (C) Distant tumors from bilateral B16F10 tumor–bearing mice were assessed by flow cytometry to determine the fraction of PD-1hiEomeshi terminally exhausted CD8+ T cells (n = 5/group) 1 week after isotype and CMP treatment. (D) IFN-γ and granzyme B expression was quantified by flow cytometry in the distant tumors and spleens 1 week after isotype or CMP treatment (n = 5/group). (E) Changes in the PD-1hi fraction of CD8+ T cells over time (n = 4/group) in isotype- and CMP-treated animals. (F) CD8+ T cell expression of TIM3, LAG3, and 2B4 at distant tumors and spleens (n = 5/group). (G) PD-1hi fraction of CD8+ T cells in treated tumors, distant tumors, spleens, and DLNs in response to treatment with CMP and each constituent agent (n = 4/group). Representative contour plots of 2 experiments are shown for the distant tumor. *P ≤ 0.05, ** P ≤ 0.01, ***P ≤ 0.001, and ****P ≤ 0.0001, by unpaired, 2-tailed Student’s t test. Given this shift away from an exhausted CD8+ T cell expression pattern toward one associated with effector CD8+ T cells, we hypothesized that CMP treatment allows APCs exposed to tumor antigens to prime tumor-specific T cells, which then accumulate within tumors to replace existing exhausted T cells. We assessed the fraction of PD-1hiEomeshiCD8+ T cells, the population of T cells associated with terminal exhaustion and resistance to PD-1 blockade (31), and found that treatment was associated with the absence of this cell population in distant tumors (Figure 5C). Functionally, we observed that CD8+ T cells in these tumors expressed higher levels of IFN-γ and granzyme B (Figure 5D). The change in the terminally exhausted T cell population was reflected in the loss of PD-1hi T cells, which became most pronounced in treated and distant tumors 1 week after the start of CMP treatment (Figure 5E) and is consistent with enhanced cytolytic function of individual T cells sorted from CMP-treated animals’ distant tumors at 1 week (Figure 3C). We next expanded our analysis to DLNs and the spleen to further assess the loss of PD-1hiCD8+ T cells outside of tumors. Interestingly, we found no similar loss of this cell population in these lymphoid organs (Figure 5E). We next examined the expression of TIM3, LAG3, and 2B4, other cell-surface markers associated with T cell exhaustion, in distant tumors and spleens. We found that all were reduced upon CMP treatment in the tumors but were either unchanged or upregulated outside of the tumors (Figure 5F). Next, we sought to determine the contribution of constituent monotherapies to the loss of exhausted PD-1hi T cells in tumors. We found that the selective absence of exhausted T cells within tumors conferred by CMP treatment could not be recapitulated with any of the respective constituents (Figure 5G). The findings with anti–PD-1 monotherapy also confirm the lack of cross-blocking between the therapeutic (RMP 1-14) and staining (RMP 1-30) anti–PD-1 antibodies (32). Finally, given the loss of intratumoral terminally exhausted T cells with CMP treatment, we hypothesized that patients with melanoma with a baseline gene expression pattern like that induced by CMP in our melanoma model would benefit most from anti–PD-1 therapy. To address this, we analyzed publicly available RNA-Seq data (ref. 33; as described in Methods) to ask whether the gene set differentially expressed in tumors after CMP treatment (Supplemental Table 1) is associated with favorable outcomes when found in pretreatment biopsies of nivolumab-treated patients. Indeed, we found that patients with such changes at baseline went on to experience significantly improved progression-free survival with nivolumab (Figure 6A). In sum, these findings suggest a model in which treatment induces the activation of BATF3+ DCs exposed to tumor antigens which then traffic to DLNs, where they prime and expand naive tumor-specific CD8+ T cells, which in turn home to tumors, replace exhausted T cell populations, and mediate tumor regression (Figure 6B). Figure 6 Differentially expressed gene set from CMP-treated animals is associated with superior outcomes in nivolumab-treated patients. (A) Progression-free survival for patients on nivolumab from the Riaz et al. cohort (n = 51) with high versus low expression of the gene set identified from differential gene expression analysis of distant tumors from mice (n = 5) treated with CMP versus isotype at 1 week (P = 0.0032, log-rank test; HR = 0.29, 95% CI = 0.11–0.69). **P ≤ 0.01, by log-rank (Mantel-Cox) test. (B) Treatment model in which intratumoral therapy induces the activation of APCs within the tumor, which then traffic to the DLNs, where new waves of antitumor T cells are primed and gain effector function. These effector T cells then circulate systemically and ultimately accumulate in local and distant tumors to replace previously exhausted T cells that had failed to control tumor growth. Discussion Having previously described the abscopal effect in the setting of radiotherapy (34) and oncolytic viral therapy (35, 36), we now show that in situ vaccination with defined nononcolytic factors can potentiate PD-1 blockade. To our knowledge, CMP is the first treatment shown to induce the absence of terminally exhausted T cells in local and distant tumors while sparing intervening lymphoid organs. At baseline, exhaustion of antitumor T cells results from the chronic TCR stimulation of a relatively small number of T cells interacting with tumor antigens. It is tempting to speculate that by expanding this pool of T cells, CMP prevents the chronic TCR stimulation that would otherwise occur. This would potentially result in the absence of exhaustion among antitumor T cells (a population enriched within tumors), whereas exhaustion among a subset of other T cells would persist in lymphoid organs. Future studies will be needed to determine the specificity of the PD-1hi T cell populations in lymphoid organs and whether they indeed represent functionally exhausted T cells. The cause of the more robust tumor control at the treated tumor compared with the distant tumor, a finding with multiple in situ vaccines (36, 37), remains unclear and is currently being investigated in our laboratory. The use of defined factors further clarifies the minimal elements necessary to induce an abscopal effect. It has been suggested that directly inducing antigen release with radiation, cryoablation, radiofrequency ablation, oncolytic viruses, and other lytic factors is critical for initiating a systemic immune response with local modalities (38). In contrast to these approaches, CMP does not directly induce antigen release. Our results thus suggest that baseline antigen release, perhaps resulting from pretreatment turnover of malignant cells, may obviate the need for intervention to release antigen. Furthermore, oncolytic modalities are associated with the introduction of diverse damage- and pathogen-associated molecular patterns. These inflammatory molecules stimulate various pattern recognition receptors. The question of which receptors are strictly necessary for inducing a systemic immune response with such modalities had thus been left open. From a clinical perspective, CMP treatment holds significant therapeutic potential for several reasons. First, toxicity may be limited, given both the low-dose intratumoral treatment and the absence of exhausted T cells selectively within tumors. Second, each of the agents in this regimen has already been used in humans (39, 40). Third, by relying on factors with well-defined targets, CMP may avoid unanticipated off-target effects. And last, by using defined factors, CMP would reduce the likelihood of limitations associated with pathogen-based (e.g., viral and bacterial) approaches including neutralizing immunity (41). While we did not conduct a systematic study to rule out toxicity associated with treatment, no discernable toxicity was observed in the treated animals apart from fur depigmentation. Since vitiligo (an analogous effect resulting from the loss of melanocytes) has been observed in a subset of patients treated with ICB, it is possible that this adverse effect may prove to be more common in humans treated with CMP. Unlike other forms of therapeutic anticancer vaccination, in situ vaccination circumvents the need to identify and synthesize antigens specific to individual patients’ tumors. In principle, when the tumor itself is used as the source of antigen for a therapeutic vaccine, treatment can be expedited, and evolving sets of tumor antigens that arise during treatment can be targeted as they emerge. Future studies will be needed to fully elucidate how other intratumoral therapies, including other TLR agonists, may impact T cell exhaustion and therapeutic efficacy in distant tumors. Models of metastatic seeding (e.g., to the liver and lung) would be of particular interest to address therapeutic efficacy in tissues with disparate immune microenvironments. Novel TLR9 agonists, for example, are thought to induce a systemic effect by activating plasmacytoid DCs (pDCs) (42–44). Whether the pDCs trigger an immune response through direct antigen presentation versus the secretion of type 1 IFN alone and how this may impact T cell exhaustion in distant tumors are questions of significant interest. On the basis of the results described here, we are currently preparing prospective clinical trials to test the CMP regimen in patients with various solid malignancies. Methods Cell lines and tumor challenge experiments. Female C57BL/6 WT and Rag1–/– mice aged 6 to 8 weeks were purchased from The Jackson Laboratory. Batf3–/– mice were a gift from L. Deng (MSKCC, New York, New York, USA), and Pmel-1 TCR–transgenic mice were a gift from N. Restifo (NCI, Bethesda, Maryland, USA). The cell lines B16F10, LLC, and Hep-55.1C were obtained from ATCC, and the MC38 line was obtained from the NCI. All cell lines were confirmed to be mycoplasma negative by the MSKCC Antibody and Bioresource Core facility. Cells were maintained in RPMI 1640 supplemented with 10% FCS, 1× nonessential amino acids, 1 mM sodium pyruvate, 2 mM l-glutamine, and penicillin with streptomycin (complete RPMI media). KPC tumor cells (1 × 106) were surgically implanted orthotopically into the tail of the pancreas 8 days prior to initial treatment, other tumors were implanted by intradermal injection of 5 × 105 cells into the flanks. Prior to treatment, mice were randomized and then treated twice weekly for 4 weeks. Tumors were measured twice weekly and monitored for at least 90 days after tumor challenge. In vivo reagent and treatments. Therapeutic in vivo mAbs anti–PD-1 (RMP1-14) and anti-CD40 (FGK45), corresponding IgG isotype controls (2A3), and depleting mAbs anti-CD4 (GK1.5) and anti-CD8+ (2.43) were purchased from Bio X Cell. RMP1-14 (250 μg) and 2A3 (250 μg) were administered intraperitoneally twice weekly. FGK45 (20 μg), MPL (5 μg), and 2A3 (20 μg) were administered concurrently intratumorally twice weekly. MPL (Sigma-Aldrich) was reconstituted as previously described (45). Depleting mAbs GK1.5 (560 μg) and 2.43 (400 μg) were administered intraperitoneally twice weekly beginning 1 day prior to treatment initiation and continued for the duration of the experiment. In vivo phagocytosis. B16F10 tumor–bearing mice were injected intratumorally with the treatments described, together with 2 × 107 FITC-conjugated 1-μm latex particles (Polysciences) as previously described (11). Twenty-four hours after treatment, DLNs were harvested and analyzed by flow cytometry. In vitro studies. For in vitro phagocytosis experiments, mice were sacrificed and bone marrow from tibiae and femurs was harvested and cultured in the presence of 20 ng/μL GM-CSF in T cell growth medium (complete RPMI media with 50 μM β-mercaptoethanol). On day 3, the supernatant was discarded and replaced with fresh media and GM-CSF. On day 6, the supernatant was discarded, and adherent cells were physically dissociated. The cells were then plated with FITC-conjugated 1-μm latex particles (Polysciences) and treated with either isotype, MPL (5 μg), or anti-CD40 (20 μg) for 24 hours before analysis by flow cytometry. For cell viability studies, when B16F10 cells reached 40% confluence, media were replaced with fresh media containing 20 μg/mL of either MPL, anti-CD40, or gemcitabine. After a 72-hour incubation, cells were assayed with the CellTiter-Glo Luminescent Cell Viability Assay (Promega) as previously described (46). Immune cell isolation and flow cytometry. Immune cells were isolated and subsequently analyzed by flow cytometry as previously described (47). Briefly, tumors and LNs were harvested by dissection from sacrificed mice and mechanically homogenized into single-cell suspensions filtered through 100-μm nylon filters (BD Biosciences) into cold RPMI supplemented with 7.5% FCS. Cell suspensions were then washed once with cold RPMI. Spleens were processed in the same way with additional RBC lysis using ACK Lysing Buffer (Lonza). All samples were resuspended in PBS with 0.5% (v/v) FCS. Samples were preincubated with anti-CD16/32 mAb (Fc block, clone 2.4G, BD Biosciences) for 15 minutes at 4°C to prevent nonspecific Fc receptor binding. For surface markers, samples were then stained for 30 minutes at 4°C with various combinations of fluorochrome-conjugated antibodies: anti–CD45–Pacific Orange (clone 30-F11, Invitrogen, Thermo Fisher Scientific); anti–CD8+-APC-Cy7 (clone 53-6.7, BD Biosciences); anti–CD4-AF700 (clone RM4-5, BD Biosciences); anti-CD11c-AF700 (clone N418, eBioscience); anti–CD86-PE (clone GL1, eBioscience); anti–PD-1–FITC (RMP1-30, eBioscience); anti–Tim-3–BV480 (clone 5D12, BD Biosciences); anti–Lag-3–APC (clone C9B7W, BioLegend); and anti–2B4-PerCP-Cy5.5 (clone 2B4, BioLegend). For intracellular stain, cells were permeabilized using a FoxP3 Fixation and Permeabilization Kit (eBioscience) and stained with various combinations of the following antibodies: anti–Ki67-PE/Dazzle 594 (clone 16A8, BioLegend); anti–Eomes-PE (clone Dan11mag, eBioscience); anti–IFN-γ-V450 (clone XMG1.2, BD Biosciences); and anti–granzyme B–PE-Cy7 (clone NGZB, eBioscience). All antibodies were purchased from BD, eBioscience, or Invitrogen (Thermo Fisher Scientific). For intracellular cytokine staining, mouse immune cells were restimulated with 500 ng/mL PMA and 1 μg/mL ionomycin in T cell growth medium (complete RPMI media with 50 μM β-mercaptoethanol) at 37°C. After 1 hour, 1× GolgiStop and 1× GolgiPlug (BD Biosciences) were added and incubated for an additional 4–6 hours at 37°C. Fc blockade, surface staining, and intracellular staining were then performed as described above. Stained cells were acquired using an LSR II Flow Cytometer or LSRFortessa X-50 and BD FACSDiva software (BD Biosciences). The data were further analyzed with FlowJo software (version 10.4). Debris and doublets were excluded on the basis of forward and side scatter measurements. Dead cells were excluded using Fixable Viability Dye eFluor 506 (eBioscience). Transcriptome profiling and gene set enrichment analysis. Bilateral B16F10 tumor–bearing B57BL/6 mice were treated as described above. Four animals per treatment group were sacrificed at 24 hours or 7 days. Bilateral tumors, DLNs, and spleens were harvested and snap-frozen in liquid nitrogen. Organs were then thawed, and total RNA was extracted with TRIzol (Invitrogen, Thermo Fisher Scientific). Expression profiling was performed using Affymetrix Clariom S and analyzed using the Transcriptome Analysis Console as previously described (48, 49). All original transcriptomics data were deposited in the NCBI’s Gene Expression Omnibus (GEO) database (GEO GSE130027). Gene set enrichment was performed using GAGE (48, 49) on the robust multichip average normalized transcript expression values to identify significant gene set enrichment from immunologic signatures in the Molecular Signatures Database (48, 49) at a FDR of less than 0.05. IFN-γ ELISPOT assay. Splenocytes were harvested from B16F10 tumor–bearing mice 1 week after initial treatment, and a single-cell suspension was generated as described above. CD8+ T cells were isolated using a CD8+ T cell Isolation Kit (Miltenyi Biotec) according to the manufacturer’s instructions. IFN-γ–producing T cells were quantified as previously described (47). Briefly, the Mouse ImmunoSpot IFN-γ Single Color ELISPOT system was used (Cellular Technology Limited). For in vitro restimulation, 1 × 105 CD8+ T cells were cocultured with 1 × 105 irradiated (60 Gy) B16F10 target cells for 16 hours. Irradiated syngeneic ID8 ovarian carcinoma target cells were used as negative control targets to assess specificity. IFN-γ spots were quantified using a ImmunoSpot S6 Micro Analyzer and ImmunoSpot Professional Software (both from Cellular Technology Limited). Killing assays. For clonogenic ex vivo killing assays (24), a 48-well tissue culture plate was filled sequentially with 5 μL PBS containing 0.1 U thrombin, 1 mg/mL human fibrinogen in 100 μL PBS, 1 mg/mL rat tail collagen I, 10% FCS, and B16F10 cells, with or without magnetic-activated cell-sorting–purified (MACS-purified) CD8+ T cells at a 100:1 ratio for splenic CD8+ T cells and 10:1 for tumor CD8+ T cells. The plates were incubated for 20 minutes at 37°C in a 95% air and 5% CO2 humidified atmosphere to allow the fibrin to gel. Gels were overlaid with 1 mL complete RPMI media with 50 μM β-mercaptoethanol and incubated at 37°C in a 95% air and 5% CO2 humidified atmosphere. Twenty-four hours later, the gels were lysed by sequential collagenase (2.5 mg/mL) and trypsin (2.5 mg/mL; Sigma-Aldrich) digestion. The lysed gels were then diluted, and the recovered melanoma cells were plated in 6-well plates for colony formation. After 7 days in culture, the plates were fixed with formaldehyde and stained with 2% methylene blue, and the colonies were counted manually. Target cell killing was calculated using the equation: 1 − (melanoma + T cells)/(melanoma alone). The killing coefficient k was calculated by applying the following equation: b = b0 e − (kp − g)t in which b is the tumor concentration at any time, b0 is the initial tumor concentration, p is the T cell concentration, k is the second-order rate constant for T cell killing of tumor, and g is the first-order rate constant for tumor growth. Adoptive transfer. CD8+ T cells were purified from spleens and LNs from Pmel-1 TCR–transgenic mice (23) using magnetic beads (Miltenyi Biotec) for positive selection as described above. Transgenic CD8+ T cells were then loaded with CellTrace Violet (CTV) (Thermo Fisher Scientific) according to the manufacturer’s instructions. On day 7 after tumor challenge, 2 × 106 CTV-labeled Pmel-1 CD8+ T cells were administered by tail-vein injection into the mice. A single treatment with CMP or isotype control was administered on day 8, and LNs were harvested on day 11. Proliferation was then assessed by flow cytometry. Multidimensional encoding of MHC multimers. Murine gp100 peptide (EGSRNQDWL, AnaSpec) was exchanged onto H2-Db monomers (courtesy of the NIH Tetramer Facility at Emory University, Atlanta, Georgia, USA) complexed with UV-cleavable peptides as previously described (50). Briefly, excess gp100 peptide was plated with UV-cleavable monomers. The monomers were destabilized by the cleavage but rescued by the presence of gp100 peptide. Monomers bearing gp100 were then conjugated via streptavidin-biotin interaction to 1 of 2 distinct fluorophores (BV786 and BB515, BD Biosciences). Distant tumors were harvested from tumor-bearing mice after 1 week of treatment. Samples were stained with 1 μg/mL multimer at 37°C for 15 minutes. Cells were rinsed with PBS with 0.5% (v/v) FCS, followed by surface staining with and 100 ng/mL BV805-labeled anti-CD8+ mAbs (BD Bioscience) for 30 minutes at 4°C. Analysis by flow cytometry on an LSR II (BD Bioscience) was performed, and the data obtained were analyzed using FlowJo software (version 10.4). Live CD8+ T cells that stained positive for both fluorophores were considered specific for gp100. . Differential gene expression and survival analyses. To correlate transcriptomic changes derived from preclinical models to patient samples, we first conducted differential gene expression analysis of our microarray data between replicates in the distant tumor at 1 week with CMP versus isotype (n = 4) using limma (version 3.38.2) (51). Genes differentially expressed at a FDR P value of 0.1 and a log fold-change of more than 0 (i.e., increased in CMP) were converted to human Entrez IDs using clusterProfiler (version 3.8.1) (52). This yielded a final gene set of 73 genes (Supplemental Table 1) for evaluation of patients with pre-therapy RNA-Seq data from the Riaz et al. anti–PD-1 cohort (n = 51) (33). Fragments per kilobase per million mapped reads (FPKMs) for patients in the Riaz et al. cohort were calculated using the “fpkm()” function in DESeq2 (version 1.22.1) (53) and publicly available scripts in GitHub (https://github.com/riazn/bms038_analysis; commit ID: 137111c). To create a binary variable for survival analysis, we calculated the mean FPKM across all genes in the gene set and stratified patients into high and low expression groups using the top quartile of the distribution of mean gene set FPKMs as a cutoff. Progression-free survival, HRs, and CIs were calculated using Survival (version 2.42.6), and statistical significance was calculated using the log-rank test. All analyses were conducted in R (version 3.5.0) (https://www.R-project.org/). Statistics. Data were analyzed for statistical significance with an unpaired, 2-tailed Student’s t test when comparing the means of 2 independent groups. All data represent the mean ± SEM. A P value of less than 0.05 was considered statistically significant. In experiments with multiple t test correction, P values were adjusted using the Holm-Sidak method. Progression-free survival of nivolumab-treated patients described here (33) was estimated using the Kaplan-Meier method. All survival data were analyzed by log-rank (Mantel-Cox) test. All experiments were repeated 2–4 times. Study approval. All mouse procedures were conducted in accordance with protocols and guidelines established at the MSKCC, and all mouse procedures and experiments were approved by the IACUC of MSKCC. Mice were maintained according to NIH animal care guidelines, under a protocol approved by the IACUC of the MSKCC. Author contributions DNK and NS designed and performed experiments, analyzed the data, and prepared the manuscript. LFC, SB, DH, RZ, RG, BG, MS, AG, and JAM designed and performed experiments as well as analyzed data. DR, RMS, CK, KSP, MC, and SH conducted bioinformatics and statistical analyses. JJH and VPB assisted with manuscript preparation. DNK, JDW, and TM oversaw the experimental design, data interpretation, and manuscript preparation. Supplemental material View Supplemental data Acknowledgments We thank the NIH Tetramer Facility for providing multimers used in this study. This research was funded in part through the NIH, NCI Cancer Center Support Grant P30 CA008748; NCI R01 CA056821; the Ludwig Collaborative and Swim Across America Laboratory; the Emerald Foundation; the Tri-Institutional Therapeutics Discovery Institute; the Memorial Sloan Kettering Technology Development Fund, MSKCC; the Parker Institute for Cancer Immunotherapy, MSKCC; the Department of Medicine, MSKCC; and Weill Cornell Medicine. RG acknowledges support from an NIH-T32 Postdoctoral Research Fellowship. Footnotes Conflict of interest: DNK, TM, and JDW are co-inventors on patent applications related to CD40 and in situ vaccination (PCT/US2016/045970), filed by MSKCC. JDW is a paid consultant for Advaxis, Bristol-Myers Squibb, Merck, Medimmune, Celgene, and Genentech and receives research funding from Bristol-Myers Squibb, Merck, Genentech, and Medimmune and honoraria from Ono Pharmaceutical Company. JDW is a cofounder of, paid consultant for, and stock option owner in Potenza Therapeutics, Tizona Therapeutics, and IMVAQ Therapeutics. JDW is a paid consultant for and has stock option ownership in BeiGene and Apricity. JDW is a paid consultant for Surface Oncology, Polaris, Polynoma, Array, Ascentage Pharma, PureTech, Chugai Pharmaceutical, FStar, Amgen, SELLAS Life Sciences, Serametrix, Neon, Eli Lilly, PsiOxus Therapeutics, Syndax Pharmaceuticals, Recepta, Amgen, and Puretech and reports stock option ownership in Adaptive Biotechnologies. JDW is an inventor on the following patents: xenogeneic DNA vaccines; alphavirus replicon particles expressing TRPTM is a consultant for Immunos Therapeutics and Pfizer. TM is a cofounder of and equity holder in IMVAQ Therapeutics. TM receives research funding from Bristol-Myers Squibb, Surface Oncology, Kyn Therapeutics, Infinity Pharmaceuticals, Peregrine Pharmaceuticals, Adaptive Biotechnologies, Leap Therapeutics, and Aprea Therapeutics. TM is an inventor on patent applications related to work on oncolytic viral therapy; alpha virus–based vaccine; neo antigen modeling; CD40, GITR, OX40, PD-1, and CTLA-4. LFC is a consultant for Merck. SB has received royalties from Agenus. RZ is an inventor on patent applications related to work on GITR, PD-1, and CTLA-JJH is a consultant for Bristol-Myers Squib, Eisai, Elly Lilly, and CytomX Therapeutics and has received research support from Bristol-Myers Squib. VPB is a recipient of an immune-oncology translational research grant from Bristol-Myers Squibb and is an inventor on a patent application related to work on neoantigen modeling. Copyright: © 2019, American Society for Clinical Investigation. Reference information: J Clin Invest. https://doi.org/10.1172/JCI128562. References Rizvi H, et al. Molecular determinants of response to anti-programmed cell death (PD)-1 and anti-programmed death-ligand 1 (PD-L1) blockade in patients with non-small-cell lung cancer profiled with targeted next-generation sequencing. J Clin Oncol. 2018;36(7):633–641. View this article via: PubMed CrossRef Google Scholar Tumeh PC, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515(7528):568–571. View this article via: PubMed CrossRef Google Scholar Pauken KE, Wherry EJ. Overcoming T cell exhaustion in infection and cancer. Trends Immunol. 2015;36(4):265–276. View this article via: PubMed CrossRef Google Scholar Hui E, et al. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science. 2017;355(6332):1428–1433. View this article via: PubMed CrossRef Google Scholar Kamphorst AO, et al. Rescue of exhausted CD8 T cells by PD-1-targeted therapies is CD28-dependent. Science. 2017;355(6332):1423–1427. View this article via: PubMed CrossRef Google Scholar Lanier LL, et al. CD80 (B7) and CD86 (B70) provide similar costimulatory signals for T cell proliferation, cytokine production, and generation of CTL. J Immunol. 1995;154(1):97–105. View this article via: PubMed Google Scholar Fend L, et al. Immune checkpoint blockade, immunogenic chemotherapy or IFN-α blockade boost the local and abscopal effects of oncolytic virotherapy. Cancer Res. 2017;77(15):4146–4157. View this article via: PubMed CrossRef Google Scholar Ager CR, Reilley MJ, Nicholas C, Bartkowiak T, Jaiswal AR, Curran MA. Intratumoral STING activation with T cell checkpoint modulation generates systemic antitumor immunity. Cancer Immunol Res. 2017;5(8):676–684. View this article via: PubMed CrossRef Google Scholar Mata-Haro V, Cekic C, Martin M, Chilton PM, Casella CR, Mitchell TC. The vaccine adjuvant monophosphoryl lipid A as a TRIF-biased agonist of TLR4. Science. 2007;316(5831):1628–1632. View this article via: PubMed CrossRef Google Scholar Vernacchio L, et al. Effect of monophosphoryl lipid A (MPL) on T-helper cells when administered as an adjuvant with pneumocococcal-CRM197 conjugate vaccine in healthy toddlers. Vaccine. 2002;20(31-32):3658–3667. View this article via: PubMed CrossRef Google Scholar Piconese S, Valzasina B, Colombo MP. OX40 triggering blocks suppression by regulatory T cells and facilitates tumor rejection. J Exp Med. 2008;205(4):825–839. View this article via: PubMed CrossRef Google Scholar Caux C, et al. Activation of human dendritic cells through CD40 cross-linking. J Exp Med. 1994;180(4):1263–1272. View this article via: PubMed CrossRef Google Scholar Guiducci C, Vicari AP, Sangaletti S, Trinchieri G, Colombo MP. Redirecting in vivo elicited tumor infiltrating macrophages and dendritic cells towards tumor rejection. Cancer Res. 2005;65(8):3437–3446. View this article via: PubMed CrossRef Google Scholar Andtbacka RH, et al. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J Clin Oncol. 2015;33(25):2780–2788. View this article via: PubMed CrossRef Google Scholar Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci U S A. 2010;107(9):4275–4280. View this article via: PubMed CrossRef Google Scholar Fu J, Malm IJ, Kadayakkara DK, Levitsky H, Pardoll D, Kim YJ. Preclinical evidence that PD1 blockade cooperates with cancer vaccine TEGVAX to elicit regression of established tumors. Cancer Res. 2014;74(15):4042–4052. View this article via: PubMed CrossRef Google Scholar Lollini PL, et al. Enhancement of experimental metastatic ability by tumor necrosis factor-alpha alone or in combination with interferon-gamma. Clin Exp Metastasis. 1990;8(2):215–224. View this article via: PubMed CrossRef Google Scholar Engelhardt JJ, et al. Marginating dendritic cells of the tumor microenvironment cross-present tumor antigens and stably engage tumor-specific T cells. Cancer Cell. 2012;21(3):402–417. View this article via: PubMed CrossRef Google Scholar Randolph GJ, Angeli V, Swartz MA. Dendritic-cell trafficking to lymph nodes through lymphatic vessels. Nat Rev Immunol. 2005;5(8):617–628. View this article via: PubMed CrossRef Google Scholar Hildner K, et al. Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science. 2008;322(5904):1097–1100. View this article via: PubMed CrossRef Google Scholar Fuertes MB, et al. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells. J Exp Med. 2011;208(10):2005–2016. View this article via: PubMed CrossRef Google Scholar Murphy TL, Tussiwand R, Murphy KM. Specificity through cooperation: BATF-IRF interactions control immune-regulatory networks. Nat Rev Immunol. 2013;13(7):499–509. View this article via: PubMed CrossRef Google Scholar Overwijk WW, et al. Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. J Exp Med. 2003;198(4):569–580. View this article via: PubMed CrossRef Google Scholar Budhu S, et al. CD8+ T cell concentration determines their efficiency in killing cognate antigen-expressing syngeneic mammalian cells in vitro and in mouse tissues. J Exp Med. 2010;207(1):223–235. View this article via: PubMed CrossRef Google Scholar Andersen RS, et al. Parallel detection of antigen-specific T cell responses by combinatorial encoding of MHC multimers. Nat Protoc. 2012;7(5):891–902. View this article via: PubMed CrossRef Google Scholar Byrne KT, Vonderheide RH. CD40 stimulation obviates innate sensors and drives T cell immunity in cancer. Cell Rep. 2016;15(12):2719–2732. View this article via: PubMed CrossRef Google Scholar Joeckel LT, et al. Mouse granzyme K has pro-inflammatory potential. Cell Death Differ. 2011;18(7):1112–1119. View this article via: PubMed CrossRef Google Scholar Wensink AC, et al. Granzyme K synergistically potentiates LPS-induced cytokine responses in human monocytes. Proc Natl Acad Sci U S A. 2014;111(16):5974–5979. View this article via: PubMed CrossRef Google Scholar Doering TA, Crawford A, Angelosanto JM, Paley MA, Ziegler CG, Wherry EJ. Network analysis reveals centrally connected genes and pathways involved in CD8+ T cell exhaustion versus memory. Immunity. 2012;37(6):1130–1144. View this article via: PubMed CrossRef Google Scholar Barber DL, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439(7077):682–687. View this article via: PubMed CrossRef Google Scholar Paley MA, et al. Progenitor and terminal subsets of CD8+ T cells cooperate to contain chronic viral infection. Science. 2012;338(6111):1220–1225. View this article via: PubMed CrossRef Google Scholar Totsuka T, et al. Regulation of murine chronic colitis by CD4+CD25- programmed death-1+ T cells. Eur J Immunol. 2005;35(6):1773–1785. View this article via: PubMed CrossRef Google Scholar Riaz N, et al. Tumor and microenvironment evolution during immunotherapy with nivolumab. Cell. 2017;171(4):934–949.e16. View this article via: PubMed CrossRef Google Scholar Postow MA, et al. Immunologic correlates of the abscopal effect in a patient with melanoma. N Engl J Med. 2012;366(10):925–931. View this article via: PubMed CrossRef Google Scholar Zamarin D, et al. Intratumoral modulation of the inducible co-stimulator ICOS by recombinant oncolytic virus promotes systemic anti-tumour immunity. Nat Commun. 2017;8:14340. View this article via: PubMed Google Scholar Zamarin D, et al. Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci Transl Med. 2014;6(226):226ra32. View this article via: PubMed CrossRef Google Scholar Dai P, et al. Intratumoral delivery of inactivated modified vaccinia virus Ankara (iMVA) induces systemic antitumor immunity via STING and Batf3-dependent dendritic cells. Sci Immunol. 2017;2(11):eaal1713. View this article via: PubMed Google Scholar Marabelle A, Tselikas L, de Baere T, Houot R. Intratumoral immunotherapy: using the tumor as the remedy. Ann Oncol. 2017;28(suppl_12):xii33–xii43. View this article via: PubMed CrossRef Google Scholar Ulrich JT, Myers KR. Monophosphoryl lipid A as an adjuvant. Past experiences and new directions. Pharm Biotechnol. 1995;6:495–524. View this article via: PubMed CrossRef Google Scholar Beatty GL, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. 2011;331(6024):1612–1616. View this article via: PubMed CrossRef Google Scholar Vidal L, et al. A phase I study of intravenous oncolytic reovirus type 3 Dearing in patients with advanced cancer. Clin Cancer Res. 2008;14(21):7127–7137. View this article via: PubMed CrossRef Google Scholar Krieg AM. Toll-like receptor 9 (TLR9) agonists in the treatment of cancer. Oncogene. 2008;27(2):161–167. View this article via: PubMed CrossRef Google Scholar Parker BS, Rautela J, Hertzog PJ. Antitumour actions of interferons: implications for cancer therapy. Nat Rev Cancer. 2016;16(3):131–144. View this article via: PubMed CrossRef Google Scholar Ribas A, et al. SD-101 in combination with pembrolizumab in advanced melanoma: results of a phase Ib, multicenter study. Cancer Discov. 2018;8(10):1250–1257. View this article via: PubMed CrossRef Google Scholar Stark R, Choi H, Koch S, Lamb F, Sherwood E. Monophosphoryl lipid A inhibits the cytokine response of endothelial cells challenged with LPS. Innate Immun. 2015;21(6):565–574. View this article via: PubMed CrossRef Google Scholar Thoreen CC, et al. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J Biol Chem. 2009;284(12):8023–8032. View this article via: PubMed CrossRef Google Scholar De Henau O, et al. Overcoming resistance to checkpoint blockade therapy by targeting PI3Kγ in myeloid cells. Nature. 2016;539(7629):443–447. View this article via: PubMed CrossRef Google Scholar Nassal DM, et al. KChIP2 is a core transcriptional regulator of cardiac excitability. Elife. 2017;:e17304. View this article via: PubMed Google Scholar Andrzejewski S, et al. PGC-1α promotes breast cancer metastasis and confers bioenergetic flexibility against metabolic drugs. Cell Metab. 2017;26(5):778–787.e5. View this article via: PubMed CrossRef Google Scholar Rodenko B, et al. Generation of peptide-MHC class I complexes through UV-mediated ligand exchange. Nat Protoc. 2006;1(3):1120–1132. View this article via: PubMed CrossRef Google Scholar Ritchie ME, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47. View this article via: PubMed CrossRef Google Scholar Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16(5):284–287. View this article via: PubMed CrossRef Google Scholar Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. View this article via: PubMed CrossRef Google Scholar Version history Version 1 (July 22, 2019): Electronic publication

Next Generation of Biosimilars and Biobetters: Challenges and Opportunities 5 hours 13 min agoComments by Editors, R&D Magazine Editor's Note: This technical paper is adapted from a webinar sponsored by Dassault Systèmes and presented by Advantage Business Marketing. The webinar featured Rakesh Dixit, PhD, as the speaker, who is an expert in the field of biologics from Medimmune and who presented his insights on the future of the transformative biologics market and helped navigate the webinar discussion. Laura Panjwani, the R&D Magazine Editor, served as the moderator.The full webcast can be viewed here: https://event.webcasts.com/starthere.jsp?ei=1192416&tp_key=1cd32f57f1 The biopharmaceutical market is a rapidly growing class of therapeutics, showing significant potential in oncology, diabetes and other disease areas. Unlike conventional chemically synthesized pharmaceuticals, biopharmaceuticals—also known as biologics—are derived from living organisms, typically using biotechnology. Examples of biologics include hormones, blood products, cytokines, monoclonal antibodies (mAbs), and vaccines, as well as gene transfer, cell therapy and tissue engineered products. There are more than 300 mAbs, more than 250 vaccines, and more than 100 other biologics— including cell and gene therapies—currently in clinical development. The global biologics market is expected to reach around $291 billion in 2020 and by 2022, 50 percent of the pharmaceutical market share is expected to be in biologics. But the future of biologics won’t be focused solely on the discovery of new therapeutics. There is also a significant market for biosimilars, biologic drugs that demonstrate high similarity to an already approved biologic reference drug, and can in turn serve as an alternative to it. Biosimilars are different than generics, which are synthetic chemical copies of their reference drugs and are identical in active ingredients, strength, dosage form and route of administration. Because biologics are made with living cells and no two molecules can be exactly the same, making a copy of a biologic is a much more involved and expensive process than creating a generic drug. There will always be very subtle differences between the biosimilar and its reference biologic. There are significant benefits to biosimilars. Biosimilars are a more cost effective way to manufacture biologics, increasing the affordability of life-saving biologics to patients. They could also increase worldwide regulatory approvals, including with the U.S. FDA. The Patient Protection and Affordable Care Act was signed into law by President Barack Obama on March 23, 2010, amending the Public Health Service Act to create an abbreviated approval pathway for biosimilars that are considered “highly similar” with an FDA-approved biological product. It is expected that by 2030 the biosimilar market will be a greater than a $240 billion opportunity, as patents on major biologics continue to expire. To be approved, biosimilars need to prove a high degree of similarity in biophysical properties, safety and effectiveness to the marketed reference product. The agent must demonstrate no meaningful difference in immunogenicity and pharmacokinetic/ pharmacodynamic (PK/PD) outcomes in a well-designed clinical trial. However, lower immunogenicity and better safety are acceptable. Sources: EvaluatePharma, Cortells Challenges facing the biosimilar market Despite its potential in cost savings, the biosimilar market still faces hurdles. There are gaps in knowledge and understanding of biosimilars among the public, and that can lead to challenges with physicians and patients’ perception of the safety and efficacy of the product. Worldwide there is also a high variability in regulations regarding the approval of biosimilars. In Europe, the regulatory pathway and litigation procedures are much clearer and more defined. In the U.S., however, there are very broad IP and patent laws, which can make patent infringements a major obstacle in effective marketing. This can impact patient access and increase cost. Health plans and pharmacy benefit managers (PBMs) can also contribute to increased costs, as some require high rebates that block biosimilar access and uptake. Finally, multiple companies also often compete to make the same biosimilar of a biological drug that has recently come off patent, causing stiff competition across the market. Overcoming these obstacles To stand out from the competition, companies that make biosimilar products can consider lowering prices or offering high rebates to PBMs. However, this approach is best suited in developing countries with high price sensitivity. In more established markets for biologics, such as the U.S., Europe and Japan, pricing alone isn’t sufficient, as there typically isn’t significant price variety among similar therapeutics. It is also possible to attempt to differentiate the molecule itself by providing better tolerability. However, regulatory agencies will not typically allow bio superiority to be claimed on a label on a biosimilar product. One potentially effective way to differentiate a biosimilar is through a novel delivery device or container closure system, which improves convenience, ease of use or patient acceptability of the therapeutic from its reference product. Some design differences in the delivery device or container closure system used with the proposed biosimilar product also may be acceptable for regulatory approval of a biosimilar. For a proposed biosimilar product in a different delivery device or container closure system, the presentation must be shown to be compatible for use with the final formulation of the biological product through appropriate studies including, for example, extractable/leachable studies and stability studies. For certain design differences in the delivery device or container closure system, performance testing and a human factors study may be needed. “Biobetters” Enhancing a biosimilar with an improved or more convenient delivery system is not the only way to differentiate a product. Highly differentiated biosuperior drugs, known as “Me-Betters” (after the term “Me-Too” drugs used to describe two highly similar therapeutics) are needed to serve patients who have increasingly become resistant to current therapies/standard of care. These “Me-Betters” or “biobetters,” are new biologics based on an existing approved biologic. Biobetters can deliver the mechanism of action of potency improvements, enhanced half-life, better safety and immunogenicity, and better and broader efficacy. These biobetters may serve high unmet medical needs more rapidly than a novel therapeutic, as they often receive shorter review and more rapid approval with better reimbursement. There are numerous biosimilars and biobetters in development today. Biobetter strategy Biopharmaceutical companies have focused on several strategies to develop biobetters. They are only working with molecules that have mechanisms of action (MOA) that are clinically proven or have a proof-of-efficacy that has been established and where addition values can be gained. They are focusing on areas where there are unmet medical needs within a known class of agents, where current drugs or their biosimilars do not already serve well. They aim to create biobetters where current agents are inadequate to treat refractory patients, relapsed patients, or those that have inconvenient dosing systems or safety concerns. To do this, they are focusing on application of best science and antibody technologies to create highly differentiated and potent biologics within the same general MOA as already established agents. Pros and cons of developing biobetters There are several positive outcomes from creating a highly differentiated biosuperior drug. Unlike with a biosimilar, there is generally no need to wait for patents to expire because all biobetters are treated as new molecular entries from a regulatory perspective. Despite these benefits, developing biobetters does come with challenges. As compared to biosimilars, the regulatory process will be longer and more expensive, as the agent is treated as an entirely new entry. As a result, clinical development cost may not be too dissimilar to innovative drug development. Biobetters also face fierce challenges for demonstrating superiority in efficacy or safety against established biologics and market leaders, and unless the benefits are superior to biosimilars, the higher costs of biobetters may be questioned. It can be complex to establish biosuperority, and not every attempt at doing so will be successful. Credit: Medimmune Credit: Medimmune One example of an extremely successful biobetter is Humira (adalimumab), an immunosuppressive drug used to treat arthritis, plaque psoriasis, ankylosing spondylitis, Crohn’s disease, and ulcerative colitis and other diseases. Humira works by binding to tumor necrosis factor-alpha (TNFα), which normally binds to TNFα receptors, leading to the inflammatory response of autoimmune diseases. Humira was the third TNF inhibitor, after infliximab and etanercept, to be approved in the U.S. A “biobetter” of the two agents, it was constructed from a fully human monoclonal antibody, which was an improvement over infliximab, a mouse-human chimeric antibody, and etanercept, a TNF receptor- IgG fusion protein. Humira resulted from collaboration between BASF Bioresearch Corporation and Cambridge Antibody Technology (CAT), which became MedImmune in 2007. The key to Humira’s discovery was the “phage display” method discovered at MedImmune. As of 2017, the drug has made more than 18 billion in the U.S. Credit: Medimmune Conclusion Biosimilars have tremendous potential to reduce the high cost of biologics and increase the affordability of biologics medicines. However, there are still barriers to the market’s success on the large scale, including patent limitations and legal challenges, a fractionated market, high discount expectations from PBMs and physician and patient acceptance. For innovation companies, highly differentiated and technologically advanced biobetters offer the greatest potential to mitigate the risks associated with the introduction of biosimilars, as they can be approved before the patent expires and have blockbuster potential. However, strategy is key, as long development time, high cost, and uncertainty in biosuperiority over existing drugs can hinder the success of a biobetter agent. RELATED READS

On June 29, 2016, the United States Patent and Trademark Office (USPTO) implemented a pilot program for expediting examination of patent applications relating to cancer immunotherapy. This pilot program was designed in support of the White House’s National Cancer Moonshot initiative to accelerate immunotherapy cancer research over the next five years. Via Dominique Blanchard
Applicants for antibody patents in the U.S. are finding it difficult to secure broad functional claims for antibodies and should be claiming structure, panelists at a biotech intellectual property conference said Nov.

Learn about the development of a transgenic rat for the generation of human monoclonal antibodies at OMT.
Open Monoclonal Technology, Inc. ("OMT") Via Krishan Maggon

Krishan Maggon 's curator insight,
March 12, 2015 10:58 AM
OMT makes the OmniAb platforms available directly to partners in a range of flexible and cost-efficient licenses. OmniAb antibody discovery and development services are also available at US-based Antibody Solutions (www.antibodysolutions.com), Europe-based Aldevron (www.aldevron.com), and China-based WuXi AppTec (www.wuxiapptec.com). Big pharma has licensed OMT tech for mabs
Pfizer, Roche, J&j, Merck Serono,
$AMGN suing $REGN, $SNY over patent infringement on three monoclonal antibody patents. Seeks #injunction: http://t.co/MswV3jlcCd

Written By: Hyeongsu Park. Edited By: Kendra Albert. The recent boom in antibody products in the pharmaceutical and biotechnology industries created the needs for a clear standard for antibody patents.
|

Antibodies constitute a staggering $145 billion annual market—an amount projected to almost double by 2026.Consequently, patents covering antibodies are among...
Gilbert C FAURE's insight:
Lens.org, an innovative tool for patents and more has been previously curated elsewhere https://www.scoop.it/topic/notebook-by-gilbertcfaure/?&tag=lens.org

KEY POINTS Pre-existing BCG immunity influences T cell responses of subunit booster vaccines. M. tuberculosis–specific subunit vaccines bypass this mechanism and improve protection. Abstract Despite the fact that the majority of people in tuberculosis (TB)–endemic areas are vaccinated with the Bacillus Calmette–Guérin (BCG) vaccine, TB remains the leading infectious cause of death. Data from both animal models and humans show that BCG and subunit vaccines induce T cells of different phenotypes, and little is known about how BCG priming influences subsequent booster vaccines. To test this, we designed a novel Mycobacterium tuberculosis–specific (or “non-BCG”) subunit vaccine with protective efficacy in both mice and guinea pigs and compared it to a known BCG boosting vaccine. In naive mice, this M. tuberculosis–specific vaccine induced similar protection compared with the BCG boosting vaccine. However, in BCG-primed animals, only the M. tuberculosis–specific vaccine added significantly to the BCG-induced protection. This correlated with the priming of T cells with a lower degree of differentiation and improved lung-homing capacity. These results have implications for TB vaccine design. This article is featured in Top Reads, p.1979 Introduction Mycobacterium tuberculosis infection is the leading cause of death because of a single infectious agent and has been in the top 10 causes of death worldwide for years (1).The only vaccine currently available against tuberculosis (TB) is M. bovis Bacillus Calmette–Guérin (BCG). When administered in early life, BCG efficiently prevents severe forms of childhood TB, but the efficacy against pulmonary disease in adulthood, the most common form of TB disease, is variable (2, 3). Thus, a new vaccine that prevents active pulmonary TB is needed to reduce M. tuberculosis transmission and TB-related mortality. CD4 T cells have been shown to be critical for host resistance to M. tuberculosis infections and are therefore the most common cell type targeted in preclinical and clinical TB vaccine development (4, 5). During M. tuberculosis infection, Ag expression and presentation have a major effect on differentiation and function of CD4 T cells. Recent mouse studies have shown that TB infection drives the differentiation of M. tuberculosis–specific CD4 T cells away from central memory T cells (e.g., secreting IL-2) toward effector/effector memory T cells that predominantly secrete IFN-γ (6). This results in a loss of self-renewing T cell subsets because of an impairment in the IL-2–producing capacity and a reduced capacity to traffic into the infected lung parenchyma (7, 8). Circulating T cells’ ability to populate the lung parenchyma has been established as a necessity for T cell–mediated protection in the lung (7, 9), possibly because a direct recognition of infected cells by CD4 T cells via the TCR is required (10). Thus, the ability to resist functional differentiation and home into inflamed lung tissues are key features for long-term protective CD4 T cells (7, 9). Similar to M. tuberculosis infection, the live mycobacterial BCG vaccine has been shown to promote differentiation and functional exhaustion of CD4 T cells in parenteral BCG-vaccinated mice, resulting in a failure to efficiently maintain long-term protection against M. tuberculosis (11–13). A recent study comparing different TB vaccines in clinical testing suggests that BCG may also induce more differentiated T cells than subunit vaccines in people (14). In line with this, we have recently shown that vaccination with an adjuvanted protein subunit vaccine (H56/CAF01) elicits less differentiated CD4 T cells with the capacity to localize to the infected parenchyma (15). In naive animals, such T cells are readily induced, but it has proven difficult to substantially reprogram the immune response after M. tuberculosis exposure by subunit vaccination (16–20). Given the similarities between the immune response arising from BCG vaccination and M. tuberculosis infection, we hypothesize that pre-existing “BCG-imprinted” T cells dictate the phenotype of the immune response induced by subsequent subunit booster vaccines. To investigate this, we designed two novel vaccines, H64 and H74, that selectively incorporated M. tuberculosis–specific (or “non-BCG”) Ags and compared the protective efficacy and T cell phenotype to a known booster vaccine sharing all of its Ags with BCG (H65) (21). Thus, H65 was tested as a classical BCG booster vaccine, whereas the H64 and H74 subunit vaccines supplement BCG’s Ag repertoire with M. tuberculosis–specific Ags. For comparability, the three vaccines consisted of six Ags that were either secreted by or associated with the type VII secretion system (also called the ESX secretion systems). H64 and H74 consisted of ESX-1–associated Ags (M. tuberculosis specific), whereas H65 consisted of Ags associated to ESX-2, 3, and 5 (also present in BCG). We first confirmed that the ESX-1 Ags were protective in mice and guinea pigs and that the level of protection was similar to the BCG boosting vaccine H65. We then moved on to show that in BCG-primed mice, the CD4 T cells induced by the H65 booster vaccine were more differentiated than the CD4 T cells induced by the ESX-1–based vaccine. Importantly, the T cells specific for the ESX-1 vaccine maintained their polyfunctionality and low differentiation status during chronic M. tuberculosis infection and were superior in entering the M. tuberculosis–infected lung parenchyma. As a result, the lung bacterial burden was significantly decreased in the BCG plus ESX-1 vaccination group compared with the groups with either BCG alone or BCG boosted with the H65 vaccine. These data add to the body of evidence supporting the use of ESX-1–associated (or other M. tuberculosis–specific/non-BCG) Ags in future TB vaccines (16, 22–25) and address the potential influence of BCG priming on subsequent booster vaccines. Materials and Methods Animals Six- to ten-week-old female mice or 400–500 g outbred female Hartley guinea pigs (Charles River Laboratories) were rested for 1 wk prior to initiation of any experimental procedures. Except for the M. tuberculosis Beijing HN878 challenge study, all mouse experiments were performed with female CB6F1 mice (Envigo) at Statens Serum Institut according to the Danish Ministry of Justice and Animal Protection Committees under permit 2014-15-2934-01065 and in compliance with European Union Directive 2010/63/EU. Mice were provided with radiation-sterilized food (Harlan Scandinavia) and water ad libitum and handled in accordance with the Danish Ministry of Justice and Animal Protection Committee regulations by authorized personnel. Infected mice were housed in a biosafety level 3 facility in cages contained within laminar flow safety enclosures (Scantainer, Scanbur). In the challenge study with the hypervirulent M. tuberculosis Beijing HN878, female C57BL/6 mice were purchased from SLC (Shizuoka, Japan). All animal experiments were performed according to the Korean Food and Drug Administration regulations and guidelines. The experimental protocols were reviewed and approved by the Ethics Committee and Institutional Animal Care and Use Committee (Permit Number: 2017-0264). All in vivo experiments were carried out under barrier conditions in an animal biological safety level 3 facility at the Avison Biomedical Research Center at Yonsei College of Medicine. Guinea pigs were maintained under animal biosafety level 3 barrier conditions in isolator cages (Thoren Caging Systems, Hazleton, PA) at Colorado State University. All experimental procedures were conducted in accordance with the Public Health Service Policy on the Humane Care and Use of Laboratory Animals and approved by the Colorado State University Institutional Animal Care and Use Committee (approval no. 13-4565A). Recombinant proteins All DNA constructs used in this study were made by chemical synthesis and codon optimized for expression in Escherichia coli before insertion into the pJ 411 expression vector (ATUM, Menlo Park, CA). Hybrid H64 and H74 were protein fusions without linkers between the six open reading frames. To minimize protein aggregation, all codons encoding cysteine were replaced with serine codons, five in H74 and three in H64. In both fusions, we added a His tag at the N-terminal end (MHHHHHH-). After transformation into E. coli BL21 (DE3) (Agilent Technologies), protein expression was induced with 1 mM isopropyl β-d-1-thiogalactopyranoside in 3-l cultures, and the proteins were purified from inclusion bodies by a three-step process as previously described (26). Hybrid H56 and H65 were designed as earlier described (21, 27) and expressed and purified in the same way as H74 and H64. The products were pure full-length products (>99% purity) with a protein concentration between 0.3 and 0.7 mg/ml and a total yield between 4 and 15 mg for the protein batches produced for this work. The identity of all purified protein batches was confirmed by mass spectrometry analysis (matrix-assisted laser desorption/ionization–time-of-flight). Immunizations and infections Mice were immunized s.c. three times at 2-wk intervals at the base of the tail with the fusion protein formulated in a cationic liposome adjuvant. Cationic liposomes (CAF01, 250 μg dimethyldioctadecyl-ammonium/50 μg trehalose 6,6-dibehenate) were emulsified with 1–10 μg fusion protein in 10 mM sterile Tris buffer (pH 7.4) to a final volume of 200 μl for each injection. Negative control mice received three equivalent doses of saline, and positive control mice received a single dose of 1 × 105 CFU M. bovis BCG Danish 1331 (Statens Serum Institut, Copenhagen, Denmark) given s.c. in the first round of immunization. In M. bovis BCG boost experiments, mice received one s.c. injection of 1 × 105 CFU M. bovis BCG Danish 1331 and were rested 8–26 wk, depending on the experiment, before vaccination three times with fusion protein as described above. Six weeks after the third immunization, mice were challenged with 50–100 M. tuberculosis strain Erdman (American Type Culture Collection), H37Rv (American Type Culture Collection 27294), Kazakhstan (mycobacterial interspersed repetitive units [MIRU] 1270-52), Vietnam (MIRU 1393-252), Beijing (MIRU 94-32), or Beijing HN878 suspended in PBS Tween 20 (0.05%). For M. tuberculosis strain Erdman, H37Rv, Beijing, Vietnam, and Kazakhstan, performed at Statens Serum Institut, we used a Biaera exposure system controlled by the AeroMP aerosol management, and for the hypervirulent M. tuberculosis strain Beijing HN878 experiment, performed at Yonsei College of Medicine, mice were infected with 60–70 virulent mycobacteria per mouse via the respiratory route using the inhalation chamber (Glas-Col, Terre Haute, IN). Guinea pigs (10/group), housed at Colorado State University, were immunized via the i.m. route and rested for 10 wk. A saline-treated and an intradermal M. bovis BCG–vaccinated group (inoculated with 103 CFU via the intradermal route) were included as a negative and positive control, respectively. Ten weeks postvaccination, guinea pigs were infected with a low-dose aerosol delivering ∼10 viable mycobacteria of virulent M. tuberculosis strain H37Rv into the lung of each animal. The animals were euthanized when they reached the set criteria established by the Institutional Animal Care and Use Committee, such as being moribund or exceeding acceptable weight loss and/or being affected in their respiratory rate (labored/heavy breathing). The body temperature was measured to track the clinical progression of the disease. For this, guinea pigs received a s.c. microchip implant (IPT-300 Bio Medic Data Systems, Seaford, DE) that allowed for the measurement of temperature and also carried information about experiment number and animal number. The body temperatures of individual guinea pigs were assessed each day at approximately the same time in the afternoon using a DAS-6006/7 scanner transponder (Bio Medic Data Systems). Isolation of cells and CFU measurements Spleen and lymph nodes from individual animals were kept at 4°C until processed through 70-μm nylon cell strainers (BD Pharmingen) followed by two washes and resuspension of the mononuclear cells in RPMI 1640 containing 5% FBS. Isolated lungs were transferred into Miltenyi C tubes containing HEPES/RPMI 1640 supplemented with collagenase (Roche/Sigma). The lungs were subsequently homogenized and digested for 30–45 min at 37°C and passed through cell strainers (BD Biosciences). After washing, the cells were resuspended in RPMI 1640 containing 5% FBS and stored at 4°C until use. For CFU measurements, lung homogenates were prepared in PBS Tween 80 (0.05%) from individual mice and plated at 3-fold serial dilutions on Middlebrook 7H11 Bacto Agar. After 3 wk of incubation at 37°C, the CFU were enumerated. Flow cytometry Single-cell suspensions of splenocytes or lung mononuclear cells (2 × 106 cells/well) were stimulated in vitro in V-bottom 96-well plates at 37°C in 200 μl complete media containing anti-CD49d (1 μg/ml) and anti-CD28 (1 μg/ml) Abs in the presence of rAg (2 μg/ml) for 1 h. Subsequently, 10 μg/ml brefeldin A (Sigma-Aldrich) was added, and the incubation continued for another 5–6 h. Following overnight storage at 4°C, cells were washed in FACS buffer (PBS containing 0.1% sodium azide and 1% FBS) and stained 30 min at 4°C for surface markers with mAbs as indicated. We used 1:400 dilutions of anti-CD4–Brilliant Violet 510 (clone RM 4.5; BioLegend), anti-CD4–Brilliant Violet 786 (clone GK1.5; BioLegend), 1:100 dilutions of anti-CD4–PerCP (clone GK1.5; BioLegend), or 1:200 dilutions of anti-CD4–FITC (clone RM4.4; BD Biosciences) and 1:600 dilutions of anti-CD44–FITC (clone IM7; eBioscience) and anti-CD8–PerCP-Cy5.5 (clone 53-6.7; eBioscience). Cells were then washed in FACS buffer, permeabilized using the Cytofix/Cytoperm kit (BD Biosciences) according to the manufacturer’s instructions, and stained intracellularly for 30 min at 4°C in dilutions of 1:200 using anti–IFN-γ–PE-Cy7 or anti–IFN-γ–PerCP-Cy5.5 (clone XMG1.2; eBioscience), anti–TNF-α–PE or anti–TNF-α–PeCy7 (clone MP6-XT22; eBioscience), anti–IL-17–allophycocyanin (clone eBio17B7; eBioscience), or dilutions of 1:100 using anti–IL-2–allophycocyanin-Cy7 (clone JES6-5H4; BD Biosciences) mAbs. Cells were subsequently washed with BD Perm/Wash Buffer (BD Biosciences) and resuspended in FACS buffer. Data were collected by running the stained cells on a FACSCanto, FACSCalibur, or FACSFortesa flow cytometer (BD Biosciences) and analyzed using FlowJo software v.10.0.7. In vivo intravascular labeling of T cells At the day of the experiment, mice were injected i.v. with 2 μg of FITC-labeled Abs against CD45.2 in a total volume of 200 μl (clone 102; BioLegend, San Diego, CA). Three minutes after Ab injection, mice were euthanized, and single-cell suspensions were prepared as described above. Adoptive transfer and lung homing For coadoptive transfer studies, donor CD4 T cells from subunit-vaccinated and M. bovis BCG plus subunit–vaccinated animals were isolated by negative selection 3 wk after the last immunization. In brief, cells were isolated from spleen, medial iliac, inguinal, and axillary lymph nodes from eight individual vaccinated donor animals, pooled within the groups and enumerated. Untouched CD4 T cell enrichment was performed from 5 × 108 cells per group using the EasySep Mouse CD4 T cell Enrichment Kit following the manufacturer’s instructions (STEMCELL Technologies). After enrichment, cells were counted, and the density was adjusted to 2.5 × 107 cells per ml for each group (93–95% purity). For tracking, the purified cells were differentially stained for 10 min with 10 μM Cell Proliferation Dye eFluor 450 or 5 μM Cell Proliferation Dye eFluor 670 (Thermo Fisher Scientific). The proliferation dyes were quenched with PBS containing 20% FBS followed by washing and resuspension in PBS. Stained cells were mixed in a ∼1:1 ratio and coadoptively transferred into recipient mice that were infected with M. tuberculosis strain Erdman 3 wk prior. Two hundred microliters (5 × 106 CD4 T cells) was injected into the lateral tail vein of individual recipient mice (the equivalent of one donor mouse per recipient mouse). Eighteen hours after transfer, recipient mice were injected with FITC-labeled anti-CD45.2 Abs for intravascular labeling (clone 102; BD Biosciences) and single-cell suspensions from the lung prepared as described above. Statistical analysis Prism 7 software (GraphPad Prism ver. 8.2.1, San Diego, CA) was used for all statistical analyses. Mean and SEM are indicated for log-transformed CFU counts. Mean and SD are indicated for immune responses. One-way ANOVA combined with Tukey multiple comparison test was used for comparing multiple groups. Statistical significant differences are indicated by asterisks in the figures: *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001. In the guinea pig experiment, the nonparametric log-rank test was used to compare the survival distributions of two samples comparing the survival curves for the vaccinated groups against the saline group. Results ESX-1–associated Ags (M. tuberculosis–specific) provide protection in mice and guinea pigs To investigate the influence of BCG priming on subsequent subunit vaccination, we first designed protective subunit vaccines that selectively incorporated M. tuberculosis–specific Ags, which are not shared with BCG. The M. tuberculosis genome encodes ∼4000 proteins, of which many are potential vaccine targets. However, ∼3900 of these have highly similar orthologs in the BCG genome, which significantly limits the number of potential Ags for this type of vaccine (28, 29). In our selection, we exploited that all BCG substrains lack the genomic locus “region of difference 1” (23), which includes the core genes for the ESX-1 secretion system. In virulent M. tuberculosis strains, ESX-1–secreted proteins are among the most immunogenic Ags and are frequently recognized in TB patients and latently infected individuals (30, 31). However, in BCG, the region of difference 1 deletion is expected to prevent priming of T cells against ESX-1–associated Ags, and we therefore selected among this group of proteins for the first subunit vaccine, referred to as H64. We selected six ESX-1–associated Ags for which proteome studies had identified the proteins in M. tuberculosis culture filtrate or membrane fractions (Table I) and constructed the H64 subunit vaccine as a recombinant fusion protein (Fig. 1A). In H64-vaccinated CB6F1 mice, the CD4 T cells recognized the EsxA, EspD, and EspR Ags (Fig. 1B), and we found that the subunit vaccine induced protection with protein doses ranging from 0.01 to 25 μg per vaccination peaking in the range of 1–5 μg (Fig. 1C). Based on this, an intermediate dose of 2 μg was selected for future mouse studies. Because disease progression and granuloma formation in M. tuberculosis–infected guinea pigs better mimic features of human TB pathology, we also tested the H64 vaccine in Hartley guinea pigs in a long-term infection model. Guinea pigs were challenged with a low dose of virulent M. tuberculosis H37Rv after vaccination with different doses of H64. Animals that reached predefined human end points (weight loss or impact on respiratory rate) were euthanized. Twenty-two weeks after being infected, all animals in the saline control group had been euthanized with a mean survival time of 16.2 wk (SD = ±1.8) (Supplemental Fig. 1A). In comparison, the mean survival time of the M. bovis BCG-vaccinated guinea pigs was 65.1 wk (±8.9). In the four H64-vaccinated groups, the mean survival time ranged from 22.4 wk (±2.3) to 41.6 wk (±9.0) (Fig. 1C). Statistical comparison confirmed that all vaccination groups were better protected than saline-vaccinated animals (p < 0.02, log-rank test). After having confirmed that the ESX-1–associated Ag combination was protective in both mice and guinea pigs, we continued our study of the H64 vaccine by testing its protective efficacy against different challenge strains. Because M. tuberculosis strains harbor genetic diversity that translates into significant differences in Ag diversity, immunogenicity, and virulence, we selected four clinically relevant M. tuberculosis strains belonging to different lineages (2–4) to ensure that the protective signal of H64 was robust and broadly relevant (Fig. 1D, Supplemental Fig. 1B) (32). Similarly to the results with M. tuberculosis Erdman in Fig. 1C, H64 vaccination induced significant protection against all four strains (p < 0.05 or lower) and was equal to or better than the protection obtained with the H56 subunit vaccine that was included to benchmark the new vaccine. In the experiment with H37Rv, H64 was more protective than BCG, but with the other clinical strains, BCG induced similar or better protection than H64 (Fig. 1D). To test how the vaccine performed against a more “aggressive” strain, we challenged mice with M. tuberculosis HN878, which is regarded as hypervirulent because of its rapid growth and induction of severe lung inflammation in mice (33). In this model, both BCG and H64 protected efficiently at week 4 of the infection (p < 0.001), but by week 12, BCG had lost most of its protection, whereas bacterial numbers in H64-vaccinated animals remained significantly lower than BCG as well as the saline control (Fig. 1E, p < 0.005 and p < 0.0001, respectively). View inlineView popup Table I. M. tuberculosis Ags in the H64 fusion protein FIGURE 1. H64 (ESX-1–associated Ags) provide protection against M. tuberculosis in mice and guinea pigs. (A) Illustration of the subunit vaccine H64. The fusion protein is based on ESX-1–associated Ags and does not share Ags with M. bovis BCG. The figure is not drawn to scale. The m.w. of the individual Ags is given in Table I. (B) Ag recognition of splenocytes after immunizing CB6F1 mice with H64 (n = 3). Single-cell cytokine expression was measured by flow cytometry. Any CD4 cell that produced either IFN-γ, TNF-α, and/or IL-2 in response to Ag stimulation was taken as Ag specific. The spleen cells were stimulated with single Ags from H64. EsxH stimulation was included as a negative control (“Control”). Bars and lines illustrate the mean and SD for each Ag. (C) Black curve: protective efficacy studies in CB6F1 mice. Animals were immunized with different doses of H64 in CAF01 adjuvant. The bacterial load was measured in lungs from individual mice 6 wk after M. tuberculosis Erdman challenge (n = 8). The number of bacteria was logarithmic transformed and subtracted from the bacteria numbers in a nonvaccinated control group. Blue curve: protective efficacy studies in guinea pigs (performed at Colorado State University). Guinea pigs were immunized with different doses of H64 in CAF01 and euthanized when predefined humane end points were met after M. tuberculosis infection. Kaplan–Meier survival curves (Supplemental Fig. 1A) were used to estimate the mean survival time for guinea pigs in each of the vaccination groups (n = 10). SDs are shown for each data point. (D) CB6F1 mice immunized with H64, H56, or M. bovis BCG or injected with saline were infected for 6 wk with one of four clinical isolates of M. tuberculosis: H37Rv, Vietnam, Nepal, and Kazakhstan (n = 6–8 per group). MIRU typing in Supplemental Fig. 1B. CFU Log10/lung ± SEM. One-way ANOVA was used for statistical comparison with the saline group for each strain; degree of freedom = 24. (E) H64 or M. bovis BCG immunized or saline-injected C57BL/6 mice were infected 6 wk after immunization with the hypervirulent M. tuberculosis strain Beijing HN878 (performed at Yonsei College of Medicine). After 4- and 12-wk infection, the bacterial burden was measured in the lung (n = 5–8). Mean values and SEMs are illustrated. One-way ANOVA was used for statistical comparison between groups for each time point; degree of freedom = 18. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. In parallel to working with H64, we designed an additional ESX-1 fusion protein in which EspF and PE35 (not immunogenic in H64) were replaced with the Ags EspB and EspA to potentially optimize immunogenicity and/or efficacy (Fig. 2A, Table II). In H74-vaccinated animals, there was a dominant CD4 T cell response to EspB and an increased recognition of EsxA and EspD (Fig. 2B). Similar to H64, H74 induced protective efficacy over a broad range of vaccination doses peaking between 1 and 5 μg (Supplemental Fig. 1C). Because both vaccines were designed to be used in BCG-primed animals, we did a direct head-to-head comparison in this setting. After challenge with M. tuberculosis Erdman, both vaccines induced robust protection on top of BCG at all the measured time points (Fig. 2C). However, at the late time point (20 wk postinfection), lung bacterial numbers were reduced by 1.7 log10 in the BCG control group (p < 0.0001), 1.88 log10 in the BCG-H64–vaccinated group, and 2.22 log10 in the BCG-H74–vaccinated group compared with the saline group. The bacterial burden was thus significantly lower in the H74-vaccinated animals (p < 0.01), and H74 was selected as the ESX-1 vaccine for further studies. FIGURE 2. Improved Ag recognition and protection of H74 compared with H64. (A) Illustration of the subunit vaccine H74. The fusion protein is based on ESX-1–associated Ags. Ags shared with H64 are in light green. The length in amino acids of the individual Ags is given in Table I. (B) Ag recognition of splenocytes isolated from H74-immunized CB6F1 mice (n = 4). The cells were stimulated with single Ags from H74, and EsxH stimulation was included as negative control (Control). CD4 T cells producing either IFN-γ, TNF-α, and/or IL-2 in response to Ag stimulation were taken as Ag specific. Means and SDs are shown for each Ag. (C) Six months after being M. bovis BCG–vaccinated, CB6F1 mice were divided into three groups and vaccinated with either H64 or H74 or saline injected. An age-matched control group was included that did not receive any of the vaccines. All animals were aerosol infected with M. tuberculosis Erdman, and the number of mycobacteria was measured in individual lungs from immunized and nonimmunized mice 6, 12, and 20 wk postinfection (n = 8 per time point). Vertical lines illustrate SEMs. One-way ANOVA was used for statistical comparison between groups at the late time point; degree of freedom = 28. **p < 0.01, ****p < 0.0001. View inlineView popup Table II. M. tuberculosis Ags in the H74 fusion protein In summary, we designed two novel vaccines based exclusively on ESX-1–associated Ags H64 and H74 which demonstrated robust protection in both mice and guinea pigs. H74 was selected for further studies in BCG-primed animals. In BCG-vaccinated mice, ESX-1–associated Ags induce less differentiated CD4 T cells and improve protection compared with BCG boosting It has been demonstrated that BCG vaccination induces highly differentiated T cells (34), but it has not been systematically investigated how this influences T cell quality and protection of subsequent subunit vaccine boosters. We approached this issue by first comparing the protective efficacy of the ESX-1–based H74 vaccine and a BCG booster vaccine (H65), described in a previous study (21). H65 consists of six EsxA family proteins related to ESX-2, -3, or -5 that are all present in BCG (Fig. 3A). Naive mice were immunized with either H65 or H74 as standalone vaccines. Six weeks after M. tuberculosis aerosol challenge, both vaccines reduced the bacterial load by more than 1.0 log10 relative to nonvaccinated animals (p < 0.0001) with no statistical difference between them (Fig. 3B). Having confirmed that the two vaccines induced similar levels of protection in naive mice, we continued by comparing their efficacy in mice that were BCG primed 6 mo prior to subunit vaccination. Twelve weeks after M. tuberculosis Erdman challenge, BCG vaccination reduced the lung bacterial number by 0.85 log10 (p < 0.05). In two separate experiments, H65 boosting did not add significantly to this protection (Fig. 3C, top, Supplemental Fig. 2A), whereas vaccination with the H74 vaccine enhanced the BCG-induced protection (p < 0.05), resulting in a 1.78 log10 reduction of the number of bacteria relative to the nonvaccinated group (p < 0.0001). This observation was robust as similar results were found in a second study with the clinical strain M. tuberculosis Kazakhstan (Fig. 3C, bottom), showing consistently that although the protection was equal in naive mice, the ESX-1 vaccine (M. tuberculosis–specific) induced better protection than the BCG boosting vaccine in BCG-primed animals. FIGURE 3. H74 vaccination (ESX-1–associated Ags) improve protection in BCG-primed animals and induce less differentiated CD4 T cells compared with BCG boosting (H65). (A) Illustration of the BCG booster vaccine H65. All six Ags are shared with M. bovis BCG (21). (B) Groups of naive CB6F1 mice were vaccinated with H74 or H65, and control groups received either a BCG vaccination or saline injections (n = 8). The bacterial numbers were enumerated in lungs 6 wk after an aerosol M. tuberculosis Erdman infection. Means and SEMs are shown by bars and lines. One-way ANOVA was used for statistical analysis; degree of freedom = 27. (C) CB6F1 mice were BCG vaccinated followed by a resting period of 6 mo before being vaccinated with either H74 or H65 (n = 7–8). The bacterial numbers were measured in individual animals 12 wk after they were infected with M. tuberculosis strain Erdman (top) or 25 wk after M. tuberculosis Kazakhstan infection (bottom). One-way ANOVA was used for statistical analysis; degree of freedom = 25 and 26. (D) Timeline for measuring T cell responses in vaccinated CB6F1 mice pre– and post–M. tuberculosis Erdman challenge in the BCG prime–boost model. (E) Splenocytes isolated from CB6F1 mice vaccinated with BCG alone or boosted with H65 were stimulated with the H65 fusion protein. In parallel, splenocytes from BCG-vaccinated animals complemented with H74 were stimulated with the H74 fusion protein. Single-cell expression of cytokine IFN-γ, TNF-α, and IL-2 was measured by flow cytometry, and the frequencies of activated (CD44high) CD4 T cells expressing any of the possible combinations of cytokines are shown in a bar plot for each vaccination group with mean and SD indicated (n = 3). Means (gray bars) and SDs (vertical lines) are shown. The pies are a simplified view of the data illustrating cytokine coexpression patterns of the specific CD4 T cells. The five identified subgroups of cytokine-producing CD4 T cells were as follows: light blue, TNF-α+; dark blue, TNF-α+ and IL-2+; green, TNF-α+, IL-2+, and IFN-γ+; orange, IFN-γ+ and TNF-α+; and red, IFN-γ+. The dotted arches illustrate the fraction of specific CD4 T cells that produced IFN-γ (red) or did not produce cytokine IFN-γ (blue) in response to ex vivo Ag stimulation. The FDS was calculated as the ratio of IFN-γ producers/IFN-γ nonproducers as previously described (6). (F) Cytokine expression profiles were measured in spleens before and in lungs after M. tuberculosis Erdman infection, and the associated FDS score was calculated for each time point and vaccination group (n = 3–4 per time point). Filled circles and vertical bars represents means and SDs. **p < 0.01, ***p < 0.001, ****p < 0.0001. Next, we compared the phenotype of the subunit-specific CD4 T cell response among the vaccinated groups before and after M. tuberculosis challenge (Fig. 3D). We used the individual CD4 T cell cytokine expression profile as a specific and sensitive measure to assess the degree of differentiation (35). For each group, we calculated a simple functional differentiation score (FDS) based on the ratio of IFN-γ producers and nonproducers as has previously been suggested (6). In BCG-vaccinated animals, we found the highest degree of T cell differentiation (FDS = 4.0) with the majority of responding CD4 T cells expressing IFN-γ either alone or in combination with TNF-α and/or IL-2 (Fig. 3E, top). In the H65-boosted group, there was almost a 3-fold increase in the percentage of H65-specific CD4 T cells compared with BCG alone, but H65 boosting induced only minor changes in the cytokine expression profile of the CD4 T cells (FDS = 2.8, Fig. 3E, middle). In contrast, H74 vaccination induced CD4 T cells with a lower degree of differentiation with almost half of the responding T cells expressing TNF-α alone or TNF-α and IL-2 in combination (FDS = 1.0, Fig. 3E, bottom). After M. tuberculosis Erdman infection, the CD4 T cells recruited to the lung maintained an FDS score of ∼ 4.0 in BCG-vaccinated mice during the initial phase of the infection. However, this increased to 10.7 after 6 wk and to 26.1 after 12 wk of infection, clearly showing a further differentiation of the T cell pool during TB infection (Fig. 3F, top). In the H65-boosted group, there was a delay in the differentiation of the CD4 T cells, but at the late time point, the FDS score had increased to 12.8 (Fig. 3F, middle). In contrast, the FDS score for the vaccine-specific CD4 T cells remained around ∼1.0 for all time points in the H74-vaccinated group (Fig. 3F, bottom). Thus, the pool of H74-specific CD4 T cells effectively resisted infection-driven differentiation throughout a 12-wk infection period. We further investigated this in a follow-up study, in which BCG-primed animals were immunized simultaneously with H74 and H65 so that each animal served as its own internal control. In these animals, the H65-specific CD4 T cells had a mean FDS of 3.0 compared with 0.84 for the H74-specific CD4 T cells, confirming that BCG boosting leads to higher T cell differentiation than vaccination with M. tuberculosis–specific Ags (Supplemental Fig. 2B). Finally, ESAT-6 has been shown to be essential for postexposure protection (16), and of relevance to the vaccination of M. tuberculosis–exposed individuals, H74 vaccination induced less differentiated T cells and lower bacterial burdens in the modified Cornell model of latent TB infection, supporting ESX-1–based vaccines for this application (27, 36) (Supplemental Fig. 2C). In summary, in BCG-primed mice, H65 vaccination did neither lead to substantial improvements in T cell differentiation nor did it add significantly to the protection induced by BCG. Conversely, vaccination with ESX-1–associated Ags (H74) induced CD4 T cells with a low differentiation score, which remained low during M. tuberculosis infection. This correlated with a significantly increased protective efficacy. In BCG-vaccinated mice, ESX-1–associated Ags induce CD4 T cells with superior lung-homing capacity Recent studies directly link T cell differentiation status to the ability to enter the infected lung parenchyma and restrict mycobacterial growth (7, 9, 15, 37). In H56/CAF01-vaccinated mice, we have previously shown that lung parenchymal CD4 T cells (protected from anti-CD45 i.v. stain) are less differentiated and express increased levels of the parenchymal homing marker, CXCR3 (15). For this study, to directly link FDS with lung parenchymal trafficking, we first confirmed that there was a strong correlation between FDS and i.v. CD45 labeling in vaccinated animals (Supplemental Fig. 3). Because our data this far showed that pre-existing BCG immunity had a major influence on the differentiation status of subunit-specific CD4 T cells, we next investigated the impact of BCG boosting on lung-homing capacity. For this, we performed an adoptive transfer experiment using donor cells from H74- and H65-vaccinated mice with or without BCG priming. Before cell transfer, we confirmed that all four groups had a solid vaccine-specific CD4 T cell response and used the cytokine expression profiles to determine the degree of CD4 T cell differentiation (Fig. 4A, 4B). Importantly, the T cell differentiation was similar after H74 and H65 vaccination when administered as standalone vaccines, confirming there was no inherent difference in the priming capability of these vaccines. However, in BCG-primed mice, H65 boosting led to a higher T cell differentiation than H74 vaccination, as previously observed (Fig. 4B). FIGURE 4. H74 (ESX-1–associated Ags) induces CD4 T cells with superior lung-homing capacity in BCG-vaccinated mice. (A) Eight weeks after being BCG vaccinated, CB6F1 mice were vaccinated with H65 or H74. Three weeks later, splenocytes were stimulated with H65 or H74 fusion protein, and the expression of cytokines IFN-γ, IL-2, and TNF-α was measured by flow cytometry. CD4 T cells that produced at least one of the cytokines in response to Ag stimulation was regarded as “Ag specific” (n = 4 per group). The mean (gray bar) and SD is shown for each group. (B) The FDS score was calculated as the ratio of IFN-γ+/IFN-γ− cells based on the cytokine expression profile for the individual animal and plotted for each of the four vaccination groups. Horizontal lines represent means, vertical lines, and SDs. One-way ANOVA was used for statistical comparison; degree of freedom = 12. (C) The influence of BCG priming on the lung-homing capacity of H65- and H74-induced CD4 T cells. CD4 T cells were purified from spleens and lymph nodes and pooled within the vaccination groups before labeling with tracker dyes to distinguish cells from donor animals with and without BCG priming. After labeling, cells were mixed 1:1 (e.g., naive plus H65:BCG plus H65) and adoptively transferred to M. tuberculosis Erdman–infected mice. The next day, intravascular localized cells in the recipient mice were labeled using FITC CD45.2, and purified lung cells were stimulated with fusion proteins to identify cytokine expressing (“Ag-specific”) CD4 T cells. For the entire gating strategy, see Supplemental Fig. 4. (D) For each of the four vaccination groups, the percentage of vaccine-specific donor cells entering into the lung parenchyma was calculated, and the influence of BCG priming on vaccine-specific homing was compared for the two subunit vaccines. The lines connect measurements from the same recipient mouse (n = 4). The statistical comparison was done by two-tailed t tests. *p < 0.05. To compare the ability of the vaccine-specific CD4 T cells to enter infected lung parenchyma, we transferred mixed populations of CD4 T cells into M. tuberculosis–infected recipients. Donor CD4 T cells from the four vaccination groups were isolated by negative selection from spleens and inguinal lymph nodes and stained either with Cell Proliferation Dye eFluor 450 or Cell Proliferation Dye eFluor 670 to distinguish cells from BCG-primed animals and cells from animals receiving the subunit vaccine as a standalone. Stained cells were mixed in a ∼1:1 ratio (subunit:BCG prime plus subunit) and cotransferred into M. tuberculosis Erdman–infected recipients in a total 5 × 106 donor CD4 T cells per recipient. Eighteen hours after transfer, recipient mice were injected with FITC-labeled anti-CD45.2 for intravascular labeling, and lung cells were harvested for flow cytometric analysis based on CDP450/670 as well as cytokine staining following H65/H74 stimulation (Fig. 4C, Supplemental Fig. 4). In mice receiving H65-specific cells, only 39.6–72.0% (mean 64%) of the cells from the BCG-H65–boosted donor mice were located in the lung parenchyma, whereas the range was 79.3–99.4% (mean 90%) for donor cells from H65-only vaccinated mice (Fig. 4D). In contrast, we found no significant differences in the percentage of H74-specific CD4 T cells in the parenchyma regardless of whether the cells came from H74 only or BCG-H74–vaccinated animals (range 78.0–99.4 and 72.0–99.4%, respectively, with means of 85.0 and 84.2%). These results clearly demonstrate that pre-existing BCG immunity significantly impacts the functionality of the T cells induced by subunit booster vaccines and that this mechanism can be efficiently bypassed by designing vaccines that selectively incorporate BCG-complementing TB Ags. Discussion The capacity of CD4 T cells to protect against M. tuberculosis is governed by their differentiation state and ability to localize to the site of infection (37). M. bovis BCG vaccination primes a polyfunctional CD4 T cell response that differentiates over time and gradually loses the ability to produce IL-2, proliferate, and to localize to the site of infection, which results in a loss of long-term protective efficacy (11–13). Heterologous prime–boost strategies, in which BCG vaccination is followed by a subunit vaccine boost, are aiming at improving the BCG-induced adaptive immunity in terms of magnitude, durability, and quality of the CD4 T cell response (38). In this study, we tested whether pre-existing BCG immunity impacts the vaccine response of either a “traditional” BCG booster vaccine (H65) or a complementary vaccine based on ESX-1–associated Ags that are specific for M. tuberculosis (H74 and H64, referred to collectively as “ESX-1 vaccines”). In the initial characterization of the ESX-1 vaccines, we demonstrated significant long-term protection in M. tuberculosis H37Rv–challenged guinea pigs as well as M. tuberculosis Erdman–challenged mice. To extend the study beyond the conventional laboratory M. tuberculosis strains, we challenged vaccinated mice with four different strains of M. tuberculosis belonging to three phylogenetically different lineages. The ESX-1 vaccine induced significant protection against all four strains, suggesting that the vaccine will be effective against a broad range of clinical isolates. Two of the selected isolates were part of the W-Beijing family of M. tuberculosis strains, one of them being the hypervirulent M. tuberculosis Beijing HN878. The Beijing strains are particularly relevant to include in this screening as they are highly prevalent, overrepresented among drug-resistant isolates (39), and significantly associated with HIV coinfection in human cases of TB meningitis (40). In a conventional protection readout at 4 wk postinfection, both the ESX-1 vaccine and BCG were highly protective against M. tuberculosis Beijing HN878. This was also true at the later time point for the ESX-1 vaccine, but in line with earlier studies (27, 41), BCG’s protective efficacy waned at the later stage of infection. In the prime–boost model, our data indicates that BCG vaccination has a major influence on the immune responses induced by subsequent subunit vaccines, depending on whether the Ags are shared with BCG or not. Specifically, we observed that boosting BCG with H65 only marginally changed the specific CD4 T cell differentiation status with little or no improvement of the protection. In contrast, BCG priming had minimal influence on the differentiation and functionality of CD4 T cells induced by the ESX-1 vaccine (not sharing Ags with BCG). The ESX-1 vaccine–specific T cells retained their differentiation status after M. tuberculosis infection, and their ability to enter the infected lung parenchyma was increased compared with the T cells in the H65-boosted group. As a result, the ESX-1 vaccine significantly augmented an already strong BCG-induced protection, and we obtained 1.8–2.9 log10 reduction in the lung bacterial load. Importantly, the BCG-priming vaccine was administered more than 6 mo prior to subunit vaccination, suggesting that it was the BCG “T cell imprint,” rather than ongoing bacterial multiplication, that influenced the response of the subunit booster vaccine. In other words, the BCG-specific CD4 T cells appeared to be “locked” with minimal capacity for reprograming into potentially more favorable phenotypes 6 mo later. This is likely because highly differentiated Th1 cells exhibit limited functional plasticity (42, 43) and that subunit boosting merely expands the existing pool (or a subset) of BCG-imprinted CD4 T cells. In addition, BCG may also induce a specific regulatory T cell response that could influence booster vaccines, although this was not investigated in this study (44, 45). Regardless, de novo priming of CD4 T cells using ESX-1–associated (or other M. tuberculosis–specific) Ags bypass this issue, which could be a useful strategy to increase durable protection in BCG-vaccinated populations. A limitation of this study was a lack of specific homing-related makers in the analysis, and in future studies, it will be important to establish whether vaccine-induced reduction of T cell differentiation leads to increased expression of such markers and/or increased contact between Ag-specific CD4 T cells and M. tuberculosis–infected macrophages in the lung. The mouse model has been extensively used to evaluate new prime–boost strategies using M. bovis BCG and subunit vaccines. The results have varied from no additional protection to almost 2 log10 protection compared with the BCG control group (46–48). It is difficult to do a comparative evaluation of these results as the studies differ with regard to vaccine design, mouse strain, M. tuberculosis challenge strain, BCG strain, and dose as well as the interval between prime–boost, boost–challenge, and challenge–sacrifice. However, by evaluating eight different available studies with BCG booster vaccines, we observed that the added protection of BCG boosting only was significant when the BCG-induced protection in itself was low (<0.5 log10) (47, 49–55). In light of our results, one explanation for this could be that a poor BCG vaccine take (or a waning response) will open up for better priming of less differentiated T cell response by the subunit vaccine. In humans, BCG will in most cases be administered to infants, and future booster vaccines are intended to be administered 10–15 y later and preferably before the protective efficacy of BCG wanes. It is not clear how pre-existing BCG immunity will influence subunit vaccination in this setting, and exposure to M. tuberculosis is also likely to play a dominant role in high-endemic areas. In this regard, results demonstrating that subunit vaccines can build on pre-existing M. tuberculosis immunity have been obtained in the recent phase IIb trial, in which M72/AS01e induced 49.7% vaccine efficacy against pulmonary disease in quantiferon positive individuals after 3 y of follow-up (56). Encouragingly, in this study, we also demonstrate that H74 vaccination significantly reduced bacterial burden in the modified Cornell model of postexposure vaccination. Similarly, regarding BCG-vaccinated quantiferon negative individuals, a recent study showed that it is possible to boost BCG protection with a subunit vaccine in adolescents and adults to some extent (57). In this study, H4:IC31 boosting led to 30.1% vaccine efficacy against sustained quantiferon conversion, and based on our data, we speculate that subunit vaccines with ESX-1–associated Ags have the potential to further improve on this result. Additionally, in the study by Nemes et al., BCG revaccination showed a vaccine efficacy of 40.5%, which has sparked renewed interest in using BCG revaccination as a readily applicable intervention (58). In such settings, the influence of recent “BCG imprinting” is likely to be significantly higher if BCG revaccination is to be combined with future subunit vaccines. In conclusion, mycobacterial priming by BCG vaccination induces highly differentiated CD4 T cells that, for at least 6 mo in the mouse model, restrict subsequent booster vaccines in priming additional protective T cells with sufficient memory and lung-homing potential. This phenomenon can efficiently be bypassed by designing vaccines with M. tuberculosis–specific Ags, like the ESX-1–associated Ags studied in this report. We suggest that future studies explore these findings in the human setting, in which it could also be investigated whether exposure to non-tuberculousis mycobacteria play a role in maintaining BCG immunity/T cell “imprint.” Disclosures C.A., N.P.H.K., I.S., E.H.K., T.L., E.M.A., M.R., I.R., P.A., and R.M. are employed by Statens Serum Institut, a nonprofit government research facility of which H56, H64, and H74 and the CAF01 adjuvant are proprietary products. C.A., P.A., and R.M. are coinventors of patents covering ESX-1–based vaccines. Acknowledgments We thank Joshua Woodworth for input on data interpretation and gratefully acknowledge Vivi Andersen and Camilla Rasmussen at Statens Serum Institut for excellent technical assistance. Footnotes This work was supported by The Danish Research Council (DFF - 7016-00310), the National Institutes of Health/National Institute of Allergy and Infectious Diseases (Grant 1R01AI135721-01), the European Union’s Horizon 2020 Framework Programme for Research and Innovation under Grant Agreement 643381 as part of the TBVAC2020 Consortium, and the National Institutes of Health/National Institute of Allergy and Infectious Diseases program Advanced Small Animal Models for the Testing of Candidate Therapeutic and Preventative Interventions against Mycobacteria (HHSN272201000009I-003, Task Order 12) at Colorado State University. The online version of this article contains supplemental material. Abbreviations used in this article: BCG Bacillus Calmette–Guérin FDS functional differentiation score MIRU mycobacterial interspersed repetitive unit TB tuberculosis. Received May 18, 2020. Accepted August 5, 2020. Copyright © 2020 by The American Association of Immunologists, Inc. This article is distributed under The American Association of Immunologists, Inc., Reuse Terms and Conditions for Author Choice articles. References ↵WHO. 2018. Global Tuberculosis Report 2018. World Health Organization, Geneva, Switzerland. ↵Trunz, B. B., P. Fine, C. Dye. 2006. Effect of BCG vaccination on childhood tuberculous meningitis and miliary tuberculosis worldwide: a meta-analysis and assessment of cost-effectiveness. Lancet 367: 1173–1180.OpenUrlCrossRefPubMed ↵Mangtani, P., I. Abubakar, C. Ariti, R. Beynon, L. Pimpin, P. E. M. Fine, L. C. Rodrigues, P. G. Smith, M. Lipman, P. F. Whiting, J. A. Sterne. 2014. Protection by BCG vaccine against tuberculosis: a systematic review of randomized controlled trials. Clin. Infect. Dis. 58: 470–480.OpenUrlCrossRefPubMed ↵Leveton, C., S. Barnass, B. Champion, S. Lucas, B. De Souza, M. Nicol, D. Banerjee, G. Rook. 1989. T-cell-mediated protection of mice against virulent Mycobacterium tuberculosis. Infect. Immun. 57: 390–395. ↵Green, A. M., R. Difazio, J. L. Flynn. 2013. IFN-γ from CD4 T cells is essential for host survival and enhances CD8 T cell function during Mycobacterium tuberculosis infection. J. Immunol. 190: 270–277. ↵Moguche, A. O., M. Musvosvi, A. Penn-Nicholson, C. R. Plumlee, H. Mearns, H. Geldenhuys, E. Smit, D. Abrahams, V. Rozot, O. Dintwe, et al. 2017. Antigen availability shapes T cell differentiation and function during tuberculosis. Cell Host Microbe 21: 695–706.e5.OpenUrlCrossRefPubMed ↵Sallin, M. A., S. Sakai, K. D. Kauffman, H. A. Young, J. Zhu, D. L. Barber. 2017. Th1 differentiation drives the accumulation of intravascular, non-protective CD4 T cells during tuberculosis. Cell Rep. 18: 3091–3104.OpenUrlCrossRef ↵He, H., P. N. Nehete, B. Nehete, E. Wieder, G. Yang, S. Buchl, K. J. Sastry. 2011. Functional impairment of central memory CD4 T cells is a potential early prognostic marker for changing viral load in SHIV-infected rhesus macaques. PLoS One 6: e19607. ↵Sakai, S., K. D. Kauffman, J. M. Schenkel, C. C. McBerry, K. D. Mayer-Barber, D. Masopust, D. L. Barber. 2014. Cutting edge: control of Mycobacterium tuberculosis infection by a subset of lung parenchyma-homing CD4 T cells. J. Immunol. 192: 2965–2969. ↵Srivastava, S., J. D. Ernst. 2013. Cutting edge: direct recognition of infected cells by CD4 T cells is required for control of intracellular Mycobacterium tuberculosis in vivo. J. Immunol. 191: 1016–1020. ↵Lindenstrøm, T., A. Moguche, M. Damborg, E. M. Agger, K. Urdahl, P. Andersen. 2018. T cells primed by live mycobacteria versus a tuberculosis subunit vaccine exhibit distinct functional properties. EBioMedicine 27: 27–39.OpenUrl Perdomo, C., U. Zedler, A. A. Kühl, L. Lozza, P. Saikali, L. E. Sander, A. Vogelzang, S. H. E. Kaufmann, A. Kupz. 2016. Mucosal BCG vaccination induces protective lung-resident memory T cell populations against tuberculosis. mBio 7: e01686-16. ↵Orme, I. M. 2010. The Achilles heel of BCG. Tuberculosis (Edinb.) 90: 329–332.OpenUrlCrossRef ↵Rodo, M. J., V. Rozot, E. Nemes, O. Dintwe, M. Hatherill, F. Little, T. J. Scriba. 2019. A comparison of antigen-specific T cell responses induced by six novel tuberculosis vaccine candidates. PLoS Pathog. 15: e1007643. ↵Woodworth, J. S., S. B. Cohen, A. O. Moguche, C. R. Plumlee, E. M. Agger, K. B. Urdahl, P. Andersen. 2017. Subunit vaccine H56/CAF01 induces a population of circulating CD4 T cells that traffic into the Mycobacterium tuberculosis-infected lung. Mucosal Immunol. 10: 555–564.OpenUrlCrossRefPubMed ↵Hoang, T., C. Aagaard, J. Dietrich, J. P. Cassidy, G. Dolganov, G. K. Schoolnik, C. V. Lundberg, E. M. Agger, P. Andersen. 2013. ESAT-6 (EsxA) and TB10.4 (EsxH) based vaccines for pre- and post-exposure tuberculosis vaccination. PLoS One 8: e80579. Billeskov, R., T. Lindenstrøm, J. Woodworth, C. Vilaplana, P. J. Cardona, J. P. Cassidy, R. Mortensen, E. M. Agger, P. Andersen. 2018. High antigen dose is detrimental to post-exposure vaccine protection against tuberculosis. Front. Immunol. 8: 1973.OpenUrl Henao-Tamayo, M., G. S. Palaniswamy, E. E. Smith, C. A. Shanley, B. Wang, I. M. Orme, R. J. Basaraba, N. M. DuTeau, D. Ordway. 2009. Post-exposure vaccination against Mycobacterium tuberculosis. Tuberculosis (Edinb.) 89: 142–148.OpenUrl Turner, J., E. R. Rhoades, M. Keen, J. T. Belisle, A. A. Frank, I. M. Orme. 2000. Effective preexposure tuberculosis vaccines fail to protect when they are given in an immunotherapeutic mode. Infect. Immun. 68: 1706–1709. ↵Taylor, J. L., O. C. Turner, R. J. Basaraba, J. T. Belisle, K. Huygen, I. M. Orme. 2003. Pulmonary necrosis resulting from DNA vaccination against tuberculosis. Infect. Immun. 71: 2192–2198. ↵Knudsen, N. P., S. Nørskov-Lauritsen, G. M. Dolganov, G. K. Schoolnik, T. Lindenstrøm, P. Andersen, E. M. Agger, C. Aagaard. 2014. Tuberculosis vaccine with high predicted population coverage and compatibility with modern diagnostics. Proc. Natl. Acad. Sci. USA 111: 1096–1101. ↵Aguilo, N., J. Gonzalo-Asensio, S. Alvarez-Arguedas, D. Marinova, A. B. Gomez, S. Uranga, R. Spallek, M. Singh, R. Audran, F. Spertini, C. Martin. 2017. Reactogenicity to major tuberculosis antigens absent in BCG is linked to improved protection against Mycobacterium tuberculosis. Nat. Commun. 8: 16085.OpenUrlCrossRef ↵Pym, A. S., P. Brodin, L. Majlessi, R. Brosch, C. Demangel, A. Williams, K. E. Griffiths, G. Marchal, C. Leclerc, S. T. Cole. 2003. Recombinant BCG exporting ESAT-6 confers enhanced protection against tuberculosis. Nat. Med. 9: 533–539.OpenUrlCrossRefPubMed Gröschel, M. I., F. Sayes, S. J. Shin, W. Frigui, A. Pawlik, M. Orgeur, R. Canetti, N. Honoré, R. Simeone, T. S. van der Werf, et al. 2017. Recombinant BCG expressing ESX-1 of Mycobacterium marinum combines low virulence with cytosolic immune signaling and improved TB protection. Cell Rep. 18: 2752–2765.OpenUrlCrossRef ↵Bottai, D., W. Frigui, S. Clark, E. Rayner, A. Zelmer, N. Andreu, M. I. de Jonge, G. J. Bancroft, A. Williams, P. Brodin, R. Brosch. 2015. Increased protective efficacy of recombinant BCG strains expressing virulence-neutral proteins of the ESX-1 secretion system. Vaccine 33: 2710–2718.OpenUrlCrossRefPubMed ↵Aagaard, C. S., T. T. Hoang, C. Vingsbo-Lundberg, J. Dietrich, P. Andersen. 2009. Quality and vaccine efficacy of CD4+ T cell responses directed to dominant and subdominant epitopes in ESAT-6 from Mycobacterium tuberculosis. J. Immunol. 183: 2659–2668. ↵Aagaard, C., T. Hoang, J. Dietrich, P. J. Cardona, A. Izzo, G. Dolganov, G. K. Schoolnik, J. P. Cassidy, R. Billeskov, P. Andersen. 2011. A multistage tuberculosis vaccine that confers efficient protection before and after exposure. Nat. Med. 17: 189–194.OpenUrlCrossRefPubMed ↵Cole, S. T., R. Brosch, J. Parkhill, T. Garnier, C. Churcher, D. Harris, S. V. Gordon, K. Eiglmeier, S. Gas, C. E. Barry III., et al. 1998. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393: 537–544.OpenUrlCrossRefPubMed ↵Behr, M. A., M. A. Wilson, W. P. Gill, H. Salamon, G. K. Schoolnik, S. Rane, P. M. Small. 1999. Comparative genomics of BCG vaccines by whole-genome DNA microarray. Science 284: 1520–1523. ↵Millington, K. A., S. M. Fortune, J. Low, A. Garces, S. M. Hingley-Wilson, M. Wickremasinghe, O. M. Kon, A. Lalvani. 2011. Rv3615c is a highly immunodominant RD1 (region of difference 1)-dependent secreted antigen specific for Mycobacterium tuberculosis infection. Proc. Natl. Acad. Sci. USA 108: 5730–5735. ↵van Pinxteren, L. A., P. Ravn, E. M. Agger, J. Pollock, P. Andersen. 2000. Diagnosis of tuberculosis based on the two specific antigens ESAT-6 and CFP10. Clin. Diagn. Lab. Immunol. 7: 155–160.OpenUrlCrossRefPubMed ↵Coscolla, M., S. Gagneux. 2010. Does M. tuberculosis genomic diversity explain disease diversity? Drug Discov. Today Dis. Mech. 7: e43–e59.OpenUrlCrossRefPubMed ↵Manca, C., L. Tsenova, S. Freeman, A. K. Barczak, M. Tovey, P. J. Murray, C. Barry, G. Kaplan. 2005. Hypervirulent M. tuberculosis W/Beijing strains upregulate type I IFNs and increase expression of negative regulators of the Jak-Stat pathway. J. Interferon Cytokine Res. 25: 694–701.OpenUrlCrossRefPubMed ↵Nandakumar, S., S. Kannanganat, J. E. Posey, R. R. Amara, S. B. Sable. 2014. Attrition of T-cell functions and simultaneous upregulation of inhibitory markers correspond with the waning of BCG-induced protection against tuberculosis in mice. PLoS One 9: e113951. ↵Seder, R. A., P. A. Darrah, M. Roederer. 2008. T-cell quality in memory and protection: implications for vaccine design. [Published erratum appears in 2008 Nat. Rev. Immunol. 8: 486.] Nat. Rev. Immunol. 8: 247–258.OpenUrlCrossRefPubMed ↵McCune, R. M. Jr.., W. McDermott, R. Tompsett. 1956. The fate of Mycobacterium tuberculosis in mouse tissues as determined by the microbial enumeration technique. II. The conversion of tuberculous infection to the latent state by the administration of pyrazinamide and a companion drug. J. Exp. Med. 104: 763–802.OpenUrlAbstract ↵Sakai, S., K. D. Mayer-Barber, D. L. Barber. 2014. Defining features of protective CD4 T cell responses to Mycobacterium tuberculosis. Curr. Opin. Immunol. 29: 137–142.OpenUrlCrossRefPubMed ↵Lewinsohn, D. A., D. M. Lewinsohn, T. J. Scriba. 2017. Polyfunctional CD4+ T cells as targets for tuberculosis vaccination. Front. Immunol. 8: 1262.OpenUrlCrossRefPubMed ↵Glynn, J. R., J. Whiteley, P. J. Bifani, K. Kremer, D. van Soolingen. 2002. Worldwide occurrence of Beijing/W strains of Mycobacterium tuberculosis: a systematic review. Emerg. Infect. Dis. 8: 843–849.OpenUrlCrossRefPubMed ↵Caws, M., G. Thwaites, K. Stepniewska, T. N. Nguyen, T. H. Nguyen, T. P. Nguyen, N. T. Mai, M. D. Phan, H. L. Tran, T. H. Tran, et al. 2006. Beijing genotype of Mycobacterium tuberculosis is significantly associated with human immunodeficiency virus infection and multidrug resistance in cases of tube

Program: Oral and Poster Abstracts Session: 704. Immunotherapies: Poster III Hematology Disease Topics & Pathways: CRS, Biological, Therapies, neurotoxicity, Adverse Events, CAR-Ts, immunotherapy, Quality Improvement Monday, December 9, 2019, 6:00 PM-8:00 PM Hall B, Level 2 (Orange County Convention Center) Martina Pennisi1,2,3*, Tania Jain, MD4*, Elena Mead, MD5*, Bianca Santomasso, MD, PhD6*, Mari Lynne Silverberg4*, Yakup Batlevi, PhD4*, Roni Shouval4,7*, Molly A. Maloy, MS1*, Sean M. Devlin, PhD8*, Connie Lee Batlevi, MD, PhD9, Renier J. Brentjens, MD, PhD10, Parastoo B. Dahi, MD 11, Claudia Diamonte, RN10*, Jessica Flynn, BS8*, Sergio A Giralt, MD4, Elizabeth Halton, NP-BC AOCNP10*, Maria Lia Palomba, MD9, Miriam Sanchez-Escamilla, MD4,12*, Craig S. Sauter, MD13, Michael Scordo, MD4, Gunjan L. Shah, MD4, Jae H. Park, MD14 and Miguel-Angel Perales, MD1,15 1Department of Medicine, Adult Bone Marrow Transplant Service, Memorial Sloan Kettering Cancer Center, New York, NY 2University of Milan, Milan, Italy 3Division of Hematology, Fondazione IRCCS Istituto Nazionale dei Tumori, Milano, Italy 4Adult Bone Marrow Transplant Service, Department of Medicine, Memorial Sloan Kettering Cancer Center, New York, NY 5Department of Anesthesiology & Critical Care Medicine, Memorial Sloan Kettering Cancer Center, New York, NY 6Brain Tumor Service, Department of Neurology, Memorial Sloan Kettering Cancer Center, New York, NY 7Hematology and BMT Division, Chaim Sheba Medical Center, Tel-Hashomer, Tel-Aviv University, Ramat-Gan, Israel 8Department of Biostatistics and Epidemiology, Memorial Sloan Kettering Cancer Center, New York, NY 9Lymphoma Service, Department of Medicine, Memorial Sloan Kettering Cancer Center, New York, NY 10Cellular Therapeutics Center, Memorial Sloan Kettering Cancer Center, New York, NY 11Department of Medicine, Adult Bone Marrow Transplant Service, Memorial Sloan-Kettering Cancer Center, New York, NY 12Department of Hematological Malignancies and Stem Cell Transplantation, Research Institute of Marques de Valdecilla (IDIVAL), Santander, Spain 13Memorial Sloan Kettering Cancer Center, New York, NY 14Leukemia Service, Department of Medicine, Memorial Sloan Kettering Cancer Center, New York, NY 15Weill Cornell Medical College, Cornell University, New York, NY Introduction Cytokine release syndrome (CRS) and immune effector cells-associated neurotoxicity syndrome (ICANS) are commonly associated with Chimeric Antigen Receptor (CAR) T cells therapy. To assess CAR T cell safety, various grading systems were developed and used in clinical trials: Lee, Penn, Memorial Sloan Kettering Cancer Center (MSKCC), CARTOX and CTCAEv5.0 for CRS; and CTCAEv4.03 and CARTOX for ICANS. While these grading systems evaluate mostly uniform symptoms, their intensity gradings differ. The American Society for Transplantation and Cellular Therapy (ASTCT) recently developed a simplified consensus grading system for CRS and ICANS. To validate the ASTCT grading, we compared it to the aforementioned gradings. Methods We included 2 populations of adult patients (pts) treated at our center: 1) B-cell acute lymphoblastic leukemia (B-ALL) treated with CD1928z CAR T cells from 2010 to 2016 (NCT01044069), and 2) diffuse large B-cell lymphoma (DLBCL) treated with axicabtagene ciloleucel (axi-cel) or tisagenlecleucel (tisa-cel) after FDA approval from 2018. Upon chart review, one expert clinician re-graded all CRS and ICANS. For validation, another expert independently graded a randomly selected sample (20%). A neurologist and an intensivist supervised the review process. CRS/ICANS rates and concordance rates were assessed for all grading systems. In pts with DLBCL (treated with axi-cel or tisa-cel), we used ASTCT grades to predict treatment according to currently available guidelines (axi-cel and tisa-cel FDA insert packages, CARTOX and NCCN guidelines) and compare it to the actual treatment received at our institution. Results We analyzed 102 pts: 53 B-ALL and 49 DLBCL (axi-cel: 36, tisa-cel: 13). According to ASTCT grading, 82% pts had CRS, 87% in B-ALL and 77% in DLBCL pts (axi-cel: 86%, tisa-cel: 54%). The concordance rate on diagnosis of CRS (yes vs no) across all scores was 99%. Concordance rate grade by grade, instead, was 27%, with major discordance in grades 1 to 3 (figure a-b). The Penn score upgraded 91% pts from gr 1 to 2 (neutropenic fever requiring inpatient antibiotics), and 93% from gr 2 to 3 (fluid responsive hypotension or low-dose oxygen). Grading concordance increased to 78% when Penn was excluded, with other differences due to: 1) CARTOX downgraded 15% of gr 2 pts with hypotension, for systolic blood pressure >90; and 2) CARTOX and Lee upgraded 8% pts for organ damage. By ASTCT, 50% pts experienced ICANS, 55% in B-ALL and 45% in DLBCL pts (axi-cel: 55%, tisa-cel: 15%). By CTCAEv4.03, ICANS incidence was 55% because 5 pts with headache and slurred speech with trouble word finding didn’t meet criteria for ICANS by ASTCT (due to normal ICE score), leading to a 91% global concordance rate. Concordance grade by grade was 57%, mainly due to gr 1 and gr 3-4 ICANS (figure c). CARTOX upgraded 42% of gr 3 pts to gr 4 because of brief generalized seizures which, notably, where mostly seen in B-ALL pts compared to DLBCL (30% vs 6%). Another pt was upgraded from gr 2 to 4 for asymptomatic intracranial pressure >20 mmHg without cerebral edema. We then looked at implications on management in the DLBCL group (Table 1). For CRS, only 4 pts with gr 3-4 CRS would receive tocilizumab according to tisa-cel’s label (doesn’t include tocilizumab for gr 2 CRS) compared to 24, 19, 25 pts according to axi-cel’s label, CARTOX and NCCN, respectively. In our practice, 22 patients received tocilizumab. For ICANS, steroids use was consistent across all guidelines (16 pts, 5 with gr 2 ICANS and 11 with gr ≥3 ICANS) and almost comparable to our practice (19 pts treated). Conversely, only a few pts at our institution received tocilizumab for ICANS with concurrent CRS (2 pts vs 7, 11, 11 for axi-cel, CARTOX and NCCN, respectively). Conclusions Over or under attribution of symptoms due to CAR T cells results in inconsistent scores across different grading systems. Current guidelines for CRS and ICANS management are based on the experience derived from single products and various grading systems, which may result in either overtreating or delaying treatment. As such, they cannot be universally applied. To avoid discrepancies in assessing safety and in managing different product toxicities, we conclude that a unified grading system should be utilized across clinical trials and in clinical practice, and that similar consensus management guidelines be developed and adopted. Disclosures: Santomasso: Juno/Celgene: Consultancy; Kite/Gilead: Consultancy; Novartis: Consultancy. Batlevi: Juno Therapeutics: Consultancy, Membership on an entity's Board of Directors or advisory committees. Brentjens: Celgene: Consultancy; JUNO Therapeutics: Consultancy, Patents & Royalties, Research Funding. Giralt: Celgene: Consultancy, Research Funding; Amgen: Consultancy, Research Funding; Actinium: Consultancy, Research Funding; Takeda: Consultancy, Research Funding; Kite: Consultancy; Jazz Pharmaceuticals: Consultancy; Johnson & Johnson: Consultancy, Research Funding; Novartis: Consultancy; Miltenyi: Research Funding; Spectrum Pharmaceuticals: Consultancy. Palomba: MSK (IP for Juno and Seres): Patents & Royalties; Evelo: Equity Ownership; Kite Pharmaceuticals: Membership on an entity's Board of Directors or advisory committees; Pharmacyclics: Membership on an entity's Board of Directors or advisory committees; Noble Insights: Consultancy; Hemedicus: Speakers Bureau; Merck & Co Inc.: Consultancy; Seres Therapeutics: Equity Ownership, Membership on an entity's Board of Directors or advisory committees; STRAXIMM: Membership on an entity's Board of Directors or advisory committees. Sauter: Precision Biosciences: Consultancy, Research Funding; Precision Biosciences: Consultancy, Research Funding; Kite/Gilead: Consultancy; Kite/Gilead: Consultancy; Celgene: Consultancy; Celgene: Consultancy; GSK: Consultancy; GSK: Consultancy; Gamida Cell: Consultancy; Gamida Cell: Consultancy; Juno Therapeutics: Consultancy, Research Funding; Juno Therapeutics: Consultancy, Research Funding; Sanofi-Genzyme: Consultancy, Research Funding; Sanofi-Genzyme: Consultancy, Research Funding; Spectrum Pharmaceuticals: Consultancy; Spectrum Pharmaceuticals: Consultancy; Novartis: Consultancy; Genmab: Consultancy; Genmab: Consultancy; Novartis: Consultancy. Scordo: Angiocrine Bioscience, Inc.: Consultancy; McKinsey & Company: Consultancy. Shah: Janssen Pharmaceutica: Research Funding; Amgen: Research Funding. Park: Allogene: Consultancy; Amgen: Consultancy; AstraZeneca: Consultancy; Autolus: Consultancy; GSK: Consultancy; Takeda: Consultancy; Novartis: Consultancy; Kite Pharma: Consultancy; Incyte: Consultancy. Perales: Miltenyi: Research Funding; Kyte/Gilead: Research Funding; Servier: Membership on an entity's Board of Directors or advisory committees; Medigene: Membership on an entity's Board of Directors or advisory committees; Merck: Consultancy, Honoraria; Takeda: Honoraria, Membership on an entity's Board of Directors or advisory committees; Omeros: Honoraria, Membership on an entity's Board of Directors or advisory committees; Novartis: Honoraria, Membership on an entity's Board of Directors or advisory committees; Nektar Therapeutics: Honoraria, Membership on an entity's Board of Directors or advisory committees; Incyte: Honoraria, Membership on an entity's Board of Directors or advisory committees, Research Funding; Bristol-Meyers Squibb: Honoraria, Membership on an entity's Board of Directors or advisory committees; Bellicum: Honoraria, Membership on an entity's Board of Directors or advisory committees; Abbvie: Honoraria, Membership on an entity's Board of Directors or advisory committees; NexImmune: Membership on an entity's Board of Directors or advisory committees; MolMed: Membership on an entity's Board of Directors or advisory committees. See more of: 704. Immunotherapies: Poster III See more of: Oral and Poster Abstracts << Previous Abstract | Next Abstract >> *signifies non-member of ASH

PubMed comprises more than 30 million citations for biomedical literature from MEDLINE, life science journals, and online books. Citations may include links to full-text content from PubMed Central and publisher web sites.

With the patents on many biological drugs soon to expire, the biosimilars revolution is about to shift into top gear. Via Krishan Maggon

As the pharmaceutical industry continues to shift toward biologic-based drugs, including monoclonal antibodies, protecting the underlying technology has been and continues to be a priority for companies. Via Krishan Maggon
Accusations of impropriety feature in escalating dispute.

Successful mid-stage trials for a migraine treatment by Alder Biopharmaceuticals show an injectable reduced patient suffering from chronic migraines.
From
www
Perspective from The New England Journal of Medicine — Progress and Hurdles for Follow-on Biologics
The challenges to achieving savings from follow-on biologics are large but not insurmountable. First, market-entry hurdles should be low enough to ensure that enough companies compete to affect prices. Public investment in technological advances that can support biosimilar development, such as advancing knowledge about glycosylating human proteins in yeast, can aid all manufacturers. The FDA can help by promulgating product-specific guidance on how companies can demonstrate biosimilarity or interchangeability, to reduce the disadvantages for the first companies to try. Legislators may also need to reexamine the process for exchanging information about potentially infringing patents, to ensure that innovator manufacturers cannot unreasonably delay the process in order to extend their market exclusivity, and to prevent biosimilar manufacturers from entering into anticompetitive settlements. Such settlements have bedeviled the generic small-molecule drug industry but, since 2003, have had to be reported to the Federal Trade Commission for evaluation of their anticompetitive effects. This requirement may have to be extended to biologic drugs. Innovative approaches will be required to ensure mandatory, rigorous postapproval research on the safety and effectiveness of biosimilars compared with their innovator predecessors in order to promote confidence in these new products. Over the long term, attention to both these areas will help ensure that U.S. patients benefit from appropriate price reductions for older biologic drugs that are essential for their clinical care. At the same time, fair but appropriately limited periods of exclusivity will reward the innovators of the original products while also spurring them to create new products rather than prolong exclusivity rights over older ones long after such monopolies should have come to a natural end. Via Krishan Maggon
Krishan Maggon 's curator insight,
May 7, 2015 2:11 AM
Perspective Progress and Hurdles for Follow-on BiologicsAmeet Sarpatwari, J.D., Ph.D., Jerry Avorn, M.D., and Aaron S. Kesselheim, M.D., J.D., M.P.H. May 6, 2015DOI: 10.1056/NEJMp1504672 Share:

L is for CLL treatments #AntibodyAtoZ Check out our review of anti-CD44 antibody for B-cell leukemia http://t.co/922YdzsVQe @DrugPatentWatch

Mersana Therapeutics has announced the USPTO granted patents that strengthen protections for Mersana's internal pipeline of Fleximer antibody-drug con (Mersana Strengthens Antibody-Drug Conjugate Intellectual Property Position with Issuance of...
|






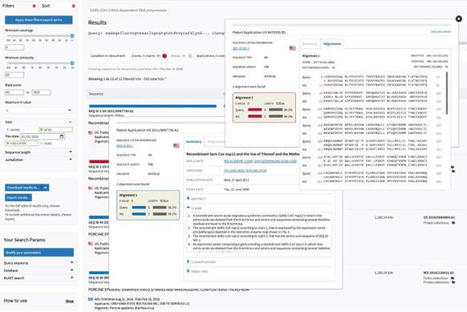


patents
https://www.scoop.it/topic/immunology-and-biotherapies?q=patents