 Your new post is loading...
Your new post is loading...

|
Scooped by
Gilbert C FAURE
January 22, 4:12 AM
|
How does BCG help the immune system fight cancer?
For decades, BCG immunotherapy has been a cornerstone treatment for high-risk non–muscle-invasive bladder cancer. Delivered directly into the bladder, it reduces tumor recurrence — yet what happens immunologically inside the patient has remained only partially understood.
In our new study, we show that :
- BCG activates BCG-specific CD4⁺ T cells,
- expands them systemically,
- and recruits them locally to the bladder, where they may contribute to tumor control.
- Remarkably, these immune cells can be detected in urine, opening new perspectives for non-invasive immune monitoring during treatment.
Pre-proof version of the article here 👇 :
https://lnkd.in/eeD_qP7N
Extremely grateful to an outstanding team, Paul Rollin, Benjamin Pluskwa, Emilie Artru, Tristan Le Vaslot, Daria Kartasheva (Ebertz), Diane Biron, Margaux Bossis, Fanny Onodi, Evanguelos Xylinas
and collaborators Joel LeMaoult, Nathalie Rouas-Freiss, Mathieu F. Chevalier, Cecilia Lindestam Arlehamn, Alessandro Sette, Dr.Biol.Sci., Alexandra Masson-Lecomte, François Desgrandchamps
#BladderCancer #Immunotherapy #BCG #CancerResearch #TranslationalImmunology Institut de Recherche Saint-Louis (IRSL/UMR 1342) Canceropôle Île-de-France, Association Francaise Urologie, Ligue contre le cancer, INSERM, UFR de Médecine Université Paris Cité
|
|
Scooped by
Gilbert C FAURE
December 30, 2020 5:36 AM
|
Click on the article title to read more.

|
Scooped by
Krishan Maggon
October 8, 2020 1:40 PM
|
KEY POINTS Pre-existing BCG immunity influences T cell responses of subunit booster vaccines. M. tuberculosis–specific subunit vaccines bypass this mechanism and improve protection. Abstract Despite the fact that the majority of people in tuberculosis (TB)–endemic areas are vaccinated with the Bacillus Calmette–Guérin (BCG) vaccine, TB remains the leading infectious cause of death. Data from both animal models and humans show that BCG and subunit vaccines induce T cells of different phenotypes, and little is known about how BCG priming influences subsequent booster vaccines. To test this, we designed a novel Mycobacterium tuberculosis–specific (or “non-BCG”) subunit vaccine with protective efficacy in both mice and guinea pigs and compared it to a known BCG boosting vaccine. In naive mice, this M. tuberculosis–specific vaccine induced similar protection compared with the BCG boosting vaccine. However, in BCG-primed animals, only the M. tuberculosis–specific vaccine added significantly to the BCG-induced protection. This correlated with the priming of T cells with a lower degree of differentiation and improved lung-homing capacity. These results have implications for TB vaccine design. This article is featured in Top Reads, p.1979 Introduction Mycobacterium tuberculosis infection is the leading cause of death because of a single infectious agent and has been in the top 10 causes of death worldwide for years (1).The only vaccine currently available against tuberculosis (TB) is M. bovis Bacillus Calmette–Guérin (BCG). When administered in early life, BCG efficiently prevents severe forms of childhood TB, but the efficacy against pulmonary disease in adulthood, the most common form of TB disease, is variable (2, 3). Thus, a new vaccine that prevents active pulmonary TB is needed to reduce M. tuberculosis transmission and TB-related mortality. CD4 T cells have been shown to be critical for host resistance to M. tuberculosis infections and are therefore the most common cell type targeted in preclinical and clinical TB vaccine development (4, 5). During M. tuberculosis infection, Ag expression and presentation have a major effect on differentiation and function of CD4 T cells. Recent mouse studies have shown that TB infection drives the differentiation of M. tuberculosis–specific CD4 T cells away from central memory T cells (e.g., secreting IL-2) toward effector/effector memory T cells that predominantly secrete IFN-γ (6). This results in a loss of self-renewing T cell subsets because of an impairment in the IL-2–producing capacity and a reduced capacity to traffic into the infected lung parenchyma (7, 8). Circulating T cells’ ability to populate the lung parenchyma has been established as a necessity for T cell–mediated protection in the lung (7, 9), possibly because a direct recognition of infected cells by CD4 T cells via the TCR is required (10). Thus, the ability to resist functional differentiation and home into inflamed lung tissues are key features for long-term protective CD4 T cells (7, 9). Similar to M. tuberculosis infection, the live mycobacterial BCG vaccine has been shown to promote differentiation and functional exhaustion of CD4 T cells in parenteral BCG-vaccinated mice, resulting in a failure to efficiently maintain long-term protection against M. tuberculosis (11–13). A recent study comparing different TB vaccines in clinical testing suggests that BCG may also induce more differentiated T cells than subunit vaccines in people (14). In line with this, we have recently shown that vaccination with an adjuvanted protein subunit vaccine (H56/CAF01) elicits less differentiated CD4 T cells with the capacity to localize to the infected parenchyma (15). In naive animals, such T cells are readily induced, but it has proven difficult to substantially reprogram the immune response after M. tuberculosis exposure by subunit vaccination (16–20). Given the similarities between the immune response arising from BCG vaccination and M. tuberculosis infection, we hypothesize that pre-existing “BCG-imprinted” T cells dictate the phenotype of the immune response induced by subsequent subunit booster vaccines. To investigate this, we designed two novel vaccines, H64 and H74, that selectively incorporated M. tuberculosis–specific (or “non-BCG”) Ags and compared the protective efficacy and T cell phenotype to a known booster vaccine sharing all of its Ags with BCG (H65) (21). Thus, H65 was tested as a classical BCG booster vaccine, whereas the H64 and H74 subunit vaccines supplement BCG’s Ag repertoire with M. tuberculosis–specific Ags. For comparability, the three vaccines consisted of six Ags that were either secreted by or associated with the type VII secretion system (also called the ESX secretion systems). H64 and H74 consisted of ESX-1–associated Ags (M. tuberculosis specific), whereas H65 consisted of Ags associated to ESX-2, 3, and 5 (also present in BCG). We first confirmed that the ESX-1 Ags were protective in mice and guinea pigs and that the level of protection was similar to the BCG boosting vaccine H65. We then moved on to show that in BCG-primed mice, the CD4 T cells induced by the H65 booster vaccine were more differentiated than the CD4 T cells induced by the ESX-1–based vaccine. Importantly, the T cells specific for the ESX-1 vaccine maintained their polyfunctionality and low differentiation status during chronic M. tuberculosis infection and were superior in entering the M. tuberculosis–infected lung parenchyma. As a result, the lung bacterial burden was significantly decreased in the BCG plus ESX-1 vaccination group compared with the groups with either BCG alone or BCG boosted with the H65 vaccine. These data add to the body of evidence supporting the use of ESX-1–associated (or other M. tuberculosis–specific/non-BCG) Ags in future TB vaccines (16, 22–25) and address the potential influence of BCG priming on subsequent booster vaccines. Materials and Methods Animals Six- to ten-week-old female mice or 400–500 g outbred female Hartley guinea pigs (Charles River Laboratories) were rested for 1 wk prior to initiation of any experimental procedures. Except for the M. tuberculosis Beijing HN878 challenge study, all mouse experiments were performed with female CB6F1 mice (Envigo) at Statens Serum Institut according to the Danish Ministry of Justice and Animal Protection Committees under permit 2014-15-2934-01065 and in compliance with European Union Directive 2010/63/EU. Mice were provided with radiation-sterilized food (Harlan Scandinavia) and water ad libitum and handled in accordance with the Danish Ministry of Justice and Animal Protection Committee regulations by authorized personnel. Infected mice were housed in a biosafety level 3 facility in cages contained within laminar flow safety enclosures (Scantainer, Scanbur). In the challenge study with the hypervirulent M. tuberculosis Beijing HN878, female C57BL/6 mice were purchased from SLC (Shizuoka, Japan). All animal experiments were performed according to the Korean Food and Drug Administration regulations and guidelines. The experimental protocols were reviewed and approved by the Ethics Committee and Institutional Animal Care and Use Committee (Permit Number: 2017-0264). All in vivo experiments were carried out under barrier conditions in an animal biological safety level 3 facility at the Avison Biomedical Research Center at Yonsei College of Medicine. Guinea pigs were maintained under animal biosafety level 3 barrier conditions in isolator cages (Thoren Caging Systems, Hazleton, PA) at Colorado State University. All experimental procedures were conducted in accordance with the Public Health Service Policy on the Humane Care and Use of Laboratory Animals and approved by the Colorado State University Institutional Animal Care and Use Committee (approval no. 13-4565A). Recombinant proteins All DNA constructs used in this study were made by chemical synthesis and codon optimized for expression in Escherichia coli before insertion into the pJ 411 expression vector (ATUM, Menlo Park, CA). Hybrid H64 and H74 were protein fusions without linkers between the six open reading frames. To minimize protein aggregation, all codons encoding cysteine were replaced with serine codons, five in H74 and three in H64. In both fusions, we added a His tag at the N-terminal end (MHHHHHH-). After transformation into E. coli BL21 (DE3) (Agilent Technologies), protein expression was induced with 1 mM isopropyl β-d-1-thiogalactopyranoside in 3-l cultures, and the proteins were purified from inclusion bodies by a three-step process as previously described (26). Hybrid H56 and H65 were designed as earlier described (21, 27) and expressed and purified in the same way as H74 and H64. The products were pure full-length products (>99% purity) with a protein concentration between 0.3 and 0.7 mg/ml and a total yield between 4 and 15 mg for the protein batches produced for this work. The identity of all purified protein batches was confirmed by mass spectrometry analysis (matrix-assisted laser desorption/ionization–time-of-flight). Immunizations and infections Mice were immunized s.c. three times at 2-wk intervals at the base of the tail with the fusion protein formulated in a cationic liposome adjuvant. Cationic liposomes (CAF01, 250 μg dimethyldioctadecyl-ammonium/50 μg trehalose 6,6-dibehenate) were emulsified with 1–10 μg fusion protein in 10 mM sterile Tris buffer (pH 7.4) to a final volume of 200 μl for each injection. Negative control mice received three equivalent doses of saline, and positive control mice received a single dose of 1 × 105 CFU M. bovis BCG Danish 1331 (Statens Serum Institut, Copenhagen, Denmark) given s.c. in the first round of immunization. In M. bovis BCG boost experiments, mice received one s.c. injection of 1 × 105 CFU M. bovis BCG Danish 1331 and were rested 8–26 wk, depending on the experiment, before vaccination three times with fusion protein as described above. Six weeks after the third immunization, mice were challenged with 50–100 M. tuberculosis strain Erdman (American Type Culture Collection), H37Rv (American Type Culture Collection 27294), Kazakhstan (mycobacterial interspersed repetitive units [MIRU] 1270-52), Vietnam (MIRU 1393-252), Beijing (MIRU 94-32), or Beijing HN878 suspended in PBS Tween 20 (0.05%). For M. tuberculosis strain Erdman, H37Rv, Beijing, Vietnam, and Kazakhstan, performed at Statens Serum Institut, we used a Biaera exposure system controlled by the AeroMP aerosol management, and for the hypervirulent M. tuberculosis strain Beijing HN878 experiment, performed at Yonsei College of Medicine, mice were infected with 60–70 virulent mycobacteria per mouse via the respiratory route using the inhalation chamber (Glas-Col, Terre Haute, IN). Guinea pigs (10/group), housed at Colorado State University, were immunized via the i.m. route and rested for 10 wk. A saline-treated and an intradermal M. bovis BCG–vaccinated group (inoculated with 103 CFU via the intradermal route) were included as a negative and positive control, respectively. Ten weeks postvaccination, guinea pigs were infected with a low-dose aerosol delivering ∼10 viable mycobacteria of virulent M. tuberculosis strain H37Rv into the lung of each animal. The animals were euthanized when they reached the set criteria established by the Institutional Animal Care and Use Committee, such as being moribund or exceeding acceptable weight loss and/or being affected in their respiratory rate (labored/heavy breathing). The body temperature was measured to track the clinical progression of the disease. For this, guinea pigs received a s.c. microchip implant (IPT-300 Bio Medic Data Systems, Seaford, DE) that allowed for the measurement of temperature and also carried information about experiment number and animal number. The body temperatures of individual guinea pigs were assessed each day at approximately the same time in the afternoon using a DAS-6006/7 scanner transponder (Bio Medic Data Systems). Isolation of cells and CFU measurements Spleen and lymph nodes from individual animals were kept at 4°C until processed through 70-μm nylon cell strainers (BD Pharmingen) followed by two washes and resuspension of the mononuclear cells in RPMI 1640 containing 5% FBS. Isolated lungs were transferred into Miltenyi C tubes containing HEPES/RPMI 1640 supplemented with collagenase (Roche/Sigma). The lungs were subsequently homogenized and digested for 30–45 min at 37°C and passed through cell strainers (BD Biosciences). After washing, the cells were resuspended in RPMI 1640 containing 5% FBS and stored at 4°C until use. For CFU measurements, lung homogenates were prepared in PBS Tween 80 (0.05%) from individual mice and plated at 3-fold serial dilutions on Middlebrook 7H11 Bacto Agar. After 3 wk of incubation at 37°C, the CFU were enumerated. Flow cytometry Single-cell suspensions of splenocytes or lung mononuclear cells (2 × 106 cells/well) were stimulated in vitro in V-bottom 96-well plates at 37°C in 200 μl complete media containing anti-CD49d (1 μg/ml) and anti-CD28 (1 μg/ml) Abs in the presence of rAg (2 μg/ml) for 1 h. Subsequently, 10 μg/ml brefeldin A (Sigma-Aldrich) was added, and the incubation continued for another 5–6 h. Following overnight storage at 4°C, cells were washed in FACS buffer (PBS containing 0.1% sodium azide and 1% FBS) and stained 30 min at 4°C for surface markers with mAbs as indicated. We used 1:400 dilutions of anti-CD4–Brilliant Violet 510 (clone RM 4.5; BioLegend), anti-CD4–Brilliant Violet 786 (clone GK1.5; BioLegend), 1:100 dilutions of anti-CD4–PerCP (clone GK1.5; BioLegend), or 1:200 dilutions of anti-CD4–FITC (clone RM4.4; BD Biosciences) and 1:600 dilutions of anti-CD44–FITC (clone IM7; eBioscience) and anti-CD8–PerCP-Cy5.5 (clone 53-6.7; eBioscience). Cells were then washed in FACS buffer, permeabilized using the Cytofix/Cytoperm kit (BD Biosciences) according to the manufacturer’s instructions, and stained intracellularly for 30 min at 4°C in dilutions of 1:200 using anti–IFN-γ–PE-Cy7 or anti–IFN-γ–PerCP-Cy5.5 (clone XMG1.2; eBioscience), anti–TNF-α–PE or anti–TNF-α–PeCy7 (clone MP6-XT22; eBioscience), anti–IL-17–allophycocyanin (clone eBio17B7; eBioscience), or dilutions of 1:100 using anti–IL-2–allophycocyanin-Cy7 (clone JES6-5H4; BD Biosciences) mAbs. Cells were subsequently washed with BD Perm/Wash Buffer (BD Biosciences) and resuspended in FACS buffer. Data were collected by running the stained cells on a FACSCanto, FACSCalibur, or FACSFortesa flow cytometer (BD Biosciences) and analyzed using FlowJo software v.10.0.7. In vivo intravascular labeling of T cells At the day of the experiment, mice were injected i.v. with 2 μg of FITC-labeled Abs against CD45.2 in a total volume of 200 μl (clone 102; BioLegend, San Diego, CA). Three minutes after Ab injection, mice were euthanized, and single-cell suspensions were prepared as described above. Adoptive transfer and lung homing For coadoptive transfer studies, donor CD4 T cells from subunit-vaccinated and M. bovis BCG plus subunit–vaccinated animals were isolated by negative selection 3 wk after the last immunization. In brief, cells were isolated from spleen, medial iliac, inguinal, and axillary lymph nodes from eight individual vaccinated donor animals, pooled within the groups and enumerated. Untouched CD4 T cell enrichment was performed from 5 × 108 cells per group using the EasySep Mouse CD4 T cell Enrichment Kit following the manufacturer’s instructions (STEMCELL Technologies). After enrichment, cells were counted, and the density was adjusted to 2.5 × 107 cells per ml for each group (93–95% purity). For tracking, the purified cells were differentially stained for 10 min with 10 μM Cell Proliferation Dye eFluor 450 or 5 μM Cell Proliferation Dye eFluor 670 (Thermo Fisher Scientific). The proliferation dyes were quenched with PBS containing 20% FBS followed by washing and resuspension in PBS. Stained cells were mixed in a ∼1:1 ratio and coadoptively transferred into recipient mice that were infected with M. tuberculosis strain Erdman 3 wk prior. Two hundred microliters (5 × 106 CD4 T cells) was injected into the lateral tail vein of individual recipient mice (the equivalent of one donor mouse per recipient mouse). Eighteen hours after transfer, recipient mice were injected with FITC-labeled anti-CD45.2 Abs for intravascular labeling (clone 102; BD Biosciences) and single-cell suspensions from the lung prepared as described above. Statistical analysis Prism 7 software (GraphPad Prism ver. 8.2.1, San Diego, CA) was used for all statistical analyses. Mean and SEM are indicated for log-transformed CFU counts. Mean and SD are indicated for immune responses. One-way ANOVA combined with Tukey multiple comparison test was used for comparing multiple groups. Statistical significant differences are indicated by asterisks in the figures: *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001. In the guinea pig experiment, the nonparametric log-rank test was used to compare the survival distributions of two samples comparing the survival curves for the vaccinated groups against the saline group. Results ESX-1–associated Ags (M. tuberculosis–specific) provide protection in mice and guinea pigs To investigate the influence of BCG priming on subsequent subunit vaccination, we first designed protective subunit vaccines that selectively incorporated M. tuberculosis–specific Ags, which are not shared with BCG. The M. tuberculosis genome encodes ∼4000 proteins, of which many are potential vaccine targets. However, ∼3900 of these have highly similar orthologs in the BCG genome, which significantly limits the number of potential Ags for this type of vaccine (28, 29). In our selection, we exploited that all BCG substrains lack the genomic locus “region of difference 1” (23), which includes the core genes for the ESX-1 secretion system. In virulent M. tuberculosis strains, ESX-1–secreted proteins are among the most immunogenic Ags and are frequently recognized in TB patients and latently infected individuals (30, 31). However, in BCG, the region of difference 1 deletion is expected to prevent priming of T cells against ESX-1–associated Ags, and we therefore selected among this group of proteins for the first subunit vaccine, referred to as H64. We selected six ESX-1–associated Ags for which proteome studies had identified the proteins in M. tuberculosis culture filtrate or membrane fractions (Table I) and constructed the H64 subunit vaccine as a recombinant fusion protein (Fig. 1A). In H64-vaccinated CB6F1 mice, the CD4 T cells recognized the EsxA, EspD, and EspR Ags (Fig. 1B), and we found that the subunit vaccine induced protection with protein doses ranging from 0.01 to 25 μg per vaccination peaking in the range of 1–5 μg (Fig. 1C). Based on this, an intermediate dose of 2 μg was selected for future mouse studies. Because disease progression and granuloma formation in M. tuberculosis–infected guinea pigs better mimic features of human TB pathology, we also tested the H64 vaccine in Hartley guinea pigs in a long-term infection model. Guinea pigs were challenged with a low dose of virulent M. tuberculosis H37Rv after vaccination with different doses of H64. Animals that reached predefined human end points (weight loss or impact on respiratory rate) were euthanized. Twenty-two weeks after being infected, all animals in the saline control group had been euthanized with a mean survival time of 16.2 wk (SD = ±1.8) (Supplemental Fig. 1A). In comparison, the mean survival time of the M. bovis BCG-vaccinated guinea pigs was 65.1 wk (±8.9). In the four H64-vaccinated groups, the mean survival time ranged from 22.4 wk (±2.3) to 41.6 wk (±9.0) (Fig. 1C). Statistical comparison confirmed that all vaccination groups were better protected than saline-vaccinated animals (p < 0.02, log-rank test). After having confirmed that the ESX-1–associated Ag combination was protective in both mice and guinea pigs, we continued our study of the H64 vaccine by testing its protective efficacy against different challenge strains. Because M. tuberculosis strains harbor genetic diversity that translates into significant differences in Ag diversity, immunogenicity, and virulence, we selected four clinically relevant M. tuberculosis strains belonging to different lineages (2–4) to ensure that the protective signal of H64 was robust and broadly relevant (Fig. 1D, Supplemental Fig. 1B) (32). Similarly to the results with M. tuberculosis Erdman in Fig. 1C, H64 vaccination induced significant protection against all four strains (p < 0.05 or lower) and was equal to or better than the protection obtained with the H56 subunit vaccine that was included to benchmark the new vaccine. In the experiment with H37Rv, H64 was more protective than BCG, but with the other clinical strains, BCG induced similar or better protection than H64 (Fig. 1D). To test how the vaccine performed against a more “aggressive” strain, we challenged mice with M. tuberculosis HN878, which is regarded as hypervirulent because of its rapid growth and induction of severe lung inflammation in mice (33). In this model, both BCG and H64 protected efficiently at week 4 of the infection (p < 0.001), but by week 12, BCG had lost most of its protection, whereas bacterial numbers in H64-vaccinated animals remained significantly lower than BCG as well as the saline control (Fig. 1E, p < 0.005 and p < 0.0001, respectively). View inlineView popup Table I. M. tuberculosis Ags in the H64 fusion protein FIGURE 1. H64 (ESX-1–associated Ags) provide protection against M. tuberculosis in mice and guinea pigs. (A) Illustration of the subunit vaccine H64. The fusion protein is based on ESX-1–associated Ags and does not share Ags with M. bovis BCG. The figure is not drawn to scale. The m.w. of the individual Ags is given in Table I. (B) Ag recognition of splenocytes after immunizing CB6F1 mice with H64 (n = 3). Single-cell cytokine expression was measured by flow cytometry. Any CD4 cell that produced either IFN-γ, TNF-α, and/or IL-2 in response to Ag stimulation was taken as Ag specific. The spleen cells were stimulated with single Ags from H64. EsxH stimulation was included as a negative control (“Control”). Bars and lines illustrate the mean and SD for each Ag. (C) Black curve: protective efficacy studies in CB6F1 mice. Animals were immunized with different doses of H64 in CAF01 adjuvant. The bacterial load was measured in lungs from individual mice 6 wk after M. tuberculosis Erdman challenge (n = 8). The number of bacteria was logarithmic transformed and subtracted from the bacteria numbers in a nonvaccinated control group. Blue curve: protective efficacy studies in guinea pigs (performed at Colorado State University). Guinea pigs were immunized with different doses of H64 in CAF01 and euthanized when predefined humane end points were met after M. tuberculosis infection. Kaplan–Meier survival curves (Supplemental Fig. 1A) were used to estimate the mean survival time for guinea pigs in each of the vaccination groups (n = 10). SDs are shown for each data point. (D) CB6F1 mice immunized with H64, H56, or M. bovis BCG or injected with saline were infected for 6 wk with one of four clinical isolates of M. tuberculosis: H37Rv, Vietnam, Nepal, and Kazakhstan (n = 6–8 per group). MIRU typing in Supplemental Fig. 1B. CFU Log10/lung ± SEM. One-way ANOVA was used for statistical comparison with the saline group for each strain; degree of freedom = 24. (E) H64 or M. bovis BCG immunized or saline-injected C57BL/6 mice were infected 6 wk after immunization with the hypervirulent M. tuberculosis strain Beijing HN878 (performed at Yonsei College of Medicine). After 4- and 12-wk infection, the bacterial burden was measured in the lung (n = 5–8). Mean values and SEMs are illustrated. One-way ANOVA was used for statistical comparison between groups for each time point; degree of freedom = 18. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. In parallel to working with H64, we designed an additional ESX-1 fusion protein in which EspF and PE35 (not immunogenic in H64) were replaced with the Ags EspB and EspA to potentially optimize immunogenicity and/or efficacy (Fig. 2A, Table II). In H74-vaccinated animals, there was a dominant CD4 T cell response to EspB and an increased recognition of EsxA and EspD (Fig. 2B). Similar to H64, H74 induced protective efficacy over a broad range of vaccination doses peaking between 1 and 5 μg (Supplemental Fig. 1C). Because both vaccines were designed to be used in BCG-primed animals, we did a direct head-to-head comparison in this setting. After challenge with M. tuberculosis Erdman, both vaccines induced robust protection on top of BCG at all the measured time points (Fig. 2C). However, at the late time point (20 wk postinfection), lung bacterial numbers were reduced by 1.7 log10 in the BCG control group (p < 0.0001), 1.88 log10 in the BCG-H64–vaccinated group, and 2.22 log10 in the BCG-H74–vaccinated group compared with the saline group. The bacterial burden was thus significantly lower in the H74-vaccinated animals (p < 0.01), and H74 was selected as the ESX-1 vaccine for further studies. FIGURE 2. Improved Ag recognition and protection of H74 compared with H64. (A) Illustration of the subunit vaccine H74. The fusion protein is based on ESX-1–associated Ags. Ags shared with H64 are in light green. The length in amino acids of the individual Ags is given in Table I. (B) Ag recognition of splenocytes isolated from H74-immunized CB6F1 mice (n = 4). The cells were stimulated with single Ags from H74, and EsxH stimulation was included as negative control (Control). CD4 T cells producing either IFN-γ, TNF-α, and/or IL-2 in response to Ag stimulation were taken as Ag specific. Means and SDs are shown for each Ag. (C) Six months after being M. bovis BCG–vaccinated, CB6F1 mice were divided into three groups and vaccinated with either H64 or H74 or saline injected. An age-matched control group was included that did not receive any of the vaccines. All animals were aerosol infected with M. tuberculosis Erdman, and the number of mycobacteria was measured in individual lungs from immunized and nonimmunized mice 6, 12, and 20 wk postinfection (n = 8 per time point). Vertical lines illustrate SEMs. One-way ANOVA was used for statistical comparison between groups at the late time point; degree of freedom = 28. **p < 0.01, ****p < 0.0001. View inlineView popup Table II. M. tuberculosis Ags in the H74 fusion protein In summary, we designed two novel vaccines based exclusively on ESX-1–associated Ags H64 and H74 which demonstrated robust protection in both mice and guinea pigs. H74 was selected for further studies in BCG-primed animals. In BCG-vaccinated mice, ESX-1–associated Ags induce less differentiated CD4 T cells and improve protection compared with BCG boosting It has been demonstrated that BCG vaccination induces highly differentiated T cells (34), but it has not been systematically investigated how this influences T cell quality and protection of subsequent subunit vaccine boosters. We approached this issue by first comparing the protective efficacy of the ESX-1–based H74 vaccine and a BCG booster vaccine (H65), described in a previous study (21). H65 consists of six EsxA family proteins related to ESX-2, -3, or -5 that are all present in BCG (Fig. 3A). Naive mice were immunized with either H65 or H74 as standalone vaccines. Six weeks after M. tuberculosis aerosol challenge, both vaccines reduced the bacterial load by more than 1.0 log10 relative to nonvaccinated animals (p < 0.0001) with no statistical difference between them (Fig. 3B). Having confirmed that the two vaccines induced similar levels of protection in naive mice, we continued by comparing their efficacy in mice that were BCG primed 6 mo prior to subunit vaccination. Twelve weeks after M. tuberculosis Erdman challenge, BCG vaccination reduced the lung bacterial number by 0.85 log10 (p < 0.05). In two separate experiments, H65 boosting did not add significantly to this protection (Fig. 3C, top, Supplemental Fig. 2A), whereas vaccination with the H74 vaccine enhanced the BCG-induced protection (p < 0.05), resulting in a 1.78 log10 reduction of the number of bacteria relative to the nonvaccinated group (p < 0.0001). This observation was robust as similar results were found in a second study with the clinical strain M. tuberculosis Kazakhstan (Fig. 3C, bottom), showing consistently that although the protection was equal in naive mice, the ESX-1 vaccine (M. tuberculosis–specific) induced better protection than the BCG boosting vaccine in BCG-primed animals. FIGURE 3. H74 vaccination (ESX-1–associated Ags) improve protection in BCG-primed animals and induce less differentiated CD4 T cells compared with BCG boosting (H65). (A) Illustration of the BCG booster vaccine H65. All six Ags are shared with M. bovis BCG (21). (B) Groups of naive CB6F1 mice were vaccinated with H74 or H65, and control groups received either a BCG vaccination or saline injections (n = 8). The bacterial numbers were enumerated in lungs 6 wk after an aerosol M. tuberculosis Erdman infection. Means and SEMs are shown by bars and lines. One-way ANOVA was used for statistical analysis; degree of freedom = 27. (C) CB6F1 mice were BCG vaccinated followed by a resting period of 6 mo before being vaccinated with either H74 or H65 (n = 7–8). The bacterial numbers were measured in individual animals 12 wk after they were infected with M. tuberculosis strain Erdman (top) or 25 wk after M. tuberculosis Kazakhstan infection (bottom). One-way ANOVA was used for statistical analysis; degree of freedom = 25 and 26. (D) Timeline for measuring T cell responses in vaccinated CB6F1 mice pre– and post–M. tuberculosis Erdman challenge in the BCG prime–boost model. (E) Splenocytes isolated from CB6F1 mice vaccinated with BCG alone or boosted with H65 were stimulated with the H65 fusion protein. In parallel, splenocytes from BCG-vaccinated animals complemented with H74 were stimulated with the H74 fusion protein. Single-cell expression of cytokine IFN-γ, TNF-α, and IL-2 was measured by flow cytometry, and the frequencies of activated (CD44high) CD4 T cells expressing any of the possible combinations of cytokines are shown in a bar plot for each vaccination group with mean and SD indicated (n = 3). Means (gray bars) and SDs (vertical lines) are shown. The pies are a simplified view of the data illustrating cytokine coexpression patterns of the specific CD4 T cells. The five identified subgroups of cytokine-producing CD4 T cells were as follows: light blue, TNF-α+; dark blue, TNF-α+ and IL-2+; green, TNF-α+, IL-2+, and IFN-γ+; orange, IFN-γ+ and TNF-α+; and red, IFN-γ+. The dotted arches illustrate the fraction of specific CD4 T cells that produced IFN-γ (red) or did not produce cytokine IFN-γ (blue) in response to ex vivo Ag stimulation. The FDS was calculated as the ratio of IFN-γ producers/IFN-γ nonproducers as previously described (6). (F) Cytokine expression profiles were measured in spleens before and in lungs after M. tuberculosis Erdman infection, and the associated FDS score was calculated for each time point and vaccination group (n = 3–4 per time point). Filled circles and vertical bars represents means and SDs. **p < 0.01, ***p < 0.001, ****p < 0.0001. Next, we compared the phenotype of the subunit-specific CD4 T cell response among the vaccinated groups before and after M. tuberculosis challenge (Fig. 3D). We used the individual CD4 T cell cytokine expression profile as a specific and sensitive measure to assess the degree of differentiation (35). For each group, we calculated a simple functional differentiation score (FDS) based on the ratio of IFN-γ producers and nonproducers as has previously been suggested (6). In BCG-vaccinated animals, we found the highest degree of T cell differentiation (FDS = 4.0) with the majority of responding CD4 T cells expressing IFN-γ either alone or in combination with TNF-α and/or IL-2 (Fig. 3E, top). In the H65-boosted group, there was almost a 3-fold increase in the percentage of H65-specific CD4 T cells compared with BCG alone, but H65 boosting induced only minor changes in the cytokine expression profile of the CD4 T cells (FDS = 2.8, Fig. 3E, middle). In contrast, H74 vaccination induced CD4 T cells with a lower degree of differentiation with almost half of the responding T cells expressing TNF-α alone or TNF-α and IL-2 in combination (FDS = 1.0, Fig. 3E, bottom). After M. tuberculosis Erdman infection, the CD4 T cells recruited to the lung maintained an FDS score of ∼ 4.0 in BCG-vaccinated mice during the initial phase of the infection. However, this increased to 10.7 after 6 wk and to 26.1 after 12 wk of infection, clearly showing a further differentiation of the T cell pool during TB infection (Fig. 3F, top). In the H65-boosted group, there was a delay in the differentiation of the CD4 T cells, but at the late time point, the FDS score had increased to 12.8 (Fig. 3F, middle). In contrast, the FDS score for the vaccine-specific CD4 T cells remained around ∼1.0 for all time points in the H74-vaccinated group (Fig. 3F, bottom). Thus, the pool of H74-specific CD4 T cells effectively resisted infection-driven differentiation throughout a 12-wk infection period. We further investigated this in a follow-up study, in which BCG-primed animals were immunized simultaneously with H74 and H65 so that each animal served as its own internal control. In these animals, the H65-specific CD4 T cells had a mean FDS of 3.0 compared with 0.84 for the H74-specific CD4 T cells, confirming that BCG boosting leads to higher T cell differentiation than vaccination with M. tuberculosis–specific Ags (Supplemental Fig. 2B). Finally, ESAT-6 has been shown to be essential for postexposure protection (16), and of relevance to the vaccination of M. tuberculosis–exposed individuals, H74 vaccination induced less differentiated T cells and lower bacterial burdens in the modified Cornell model of latent TB infection, supporting ESX-1–based vaccines for this application (27, 36) (Supplemental Fig. 2C). In summary, in BCG-primed mice, H65 vaccination did neither lead to substantial improvements in T cell differentiation nor did it add significantly to the protection induced by BCG. Conversely, vaccination with ESX-1–associated Ags (H74) induced CD4 T cells with a low differentiation score, which remained low during M. tuberculosis infection. This correlated with a significantly increased protective efficacy. In BCG-vaccinated mice, ESX-1–associated Ags induce CD4 T cells with superior lung-homing capacity Recent studies directly link T cell differentiation status to the ability to enter the infected lung parenchyma and restrict mycobacterial growth (7, 9, 15, 37). In H56/CAF01-vaccinated mice, we have previously shown that lung parenchymal CD4 T cells (protected from anti-CD45 i.v. stain) are less differentiated and express increased levels of the parenchymal homing marker, CXCR3 (15). For this study, to directly link FDS with lung parenchymal trafficking, we first confirmed that there was a strong correlation between FDS and i.v. CD45 labeling in vaccinated animals (Supplemental Fig. 3). Because our data this far showed that pre-existing BCG immunity had a major influence on the differentiation status of subunit-specific CD4 T cells, we next investigated the impact of BCG boosting on lung-homing capacity. For this, we performed an adoptive transfer experiment using donor cells from H74- and H65-vaccinated mice with or without BCG priming. Before cell transfer, we confirmed that all four groups had a solid vaccine-specific CD4 T cell response and used the cytokine expression profiles to determine the degree of CD4 T cell differentiation (Fig. 4A, 4B). Importantly, the T cell differentiation was similar after H74 and H65 vaccination when administered as standalone vaccines, confirming there was no inherent difference in the priming capability of these vaccines. However, in BCG-primed mice, H65 boosting led to a higher T cell differentiation than H74 vaccination, as previously observed (Fig. 4B). FIGURE 4. H74 (ESX-1–associated Ags) induces CD4 T cells with superior lung-homing capacity in BCG-vaccinated mice. (A) Eight weeks after being BCG vaccinated, CB6F1 mice were vaccinated with H65 or H74. Three weeks later, splenocytes were stimulated with H65 or H74 fusion protein, and the expression of cytokines IFN-γ, IL-2, and TNF-α was measured by flow cytometry. CD4 T cells that produced at least one of the cytokines in response to Ag stimulation was regarded as “Ag specific” (n = 4 per group). The mean (gray bar) and SD is shown for each group. (B) The FDS score was calculated as the ratio of IFN-γ+/IFN-γ− cells based on the cytokine expression profile for the individual animal and plotted for each of the four vaccination groups. Horizontal lines represent means, vertical lines, and SDs. One-way ANOVA was used for statistical comparison; degree of freedom = 12. (C) The influence of BCG priming on the lung-homing capacity of H65- and H74-induced CD4 T cells. CD4 T cells were purified from spleens and lymph nodes and pooled within the vaccination groups before labeling with tracker dyes to distinguish cells from donor animals with and without BCG priming. After labeling, cells were mixed 1:1 (e.g., naive plus H65:BCG plus H65) and adoptively transferred to M. tuberculosis Erdman–infected mice. The next day, intravascular localized cells in the recipient mice were labeled using FITC CD45.2, and purified lung cells were stimulated with fusion proteins to identify cytokine expressing (“Ag-specific”) CD4 T cells. For the entire gating strategy, see Supplemental Fig. 4. (D) For each of the four vaccination groups, the percentage of vaccine-specific donor cells entering into the lung parenchyma was calculated, and the influence of BCG priming on vaccine-specific homing was compared for the two subunit vaccines. The lines connect measurements from the same recipient mouse (n = 4). The statistical comparison was done by two-tailed t tests. *p < 0.05. To compare the ability of the vaccine-specific CD4 T cells to enter infected lung parenchyma, we transferred mixed populations of CD4 T cells into M. tuberculosis–infected recipients. Donor CD4 T cells from the four vaccination groups were isolated by negative selection from spleens and inguinal lymph nodes and stained either with Cell Proliferation Dye eFluor 450 or Cell Proliferation Dye eFluor 670 to distinguish cells from BCG-primed animals and cells from animals receiving the subunit vaccine as a standalone. Stained cells were mixed in a ∼1:1 ratio (subunit:BCG prime plus subunit) and cotransferred into M. tuberculosis Erdman–infected recipients in a total 5 × 106 donor CD4 T cells per recipient. Eighteen hours after transfer, recipient mice were injected with FITC-labeled anti-CD45.2 for intravascular labeling, and lung cells were harvested for flow cytometric analysis based on CDP450/670 as well as cytokine staining following H65/H74 stimulation (Fig. 4C, Supplemental Fig. 4). In mice receiving H65-specific cells, only 39.6–72.0% (mean 64%) of the cells from the BCG-H65–boosted donor mice were located in the lung parenchyma, whereas the range was 79.3–99.4% (mean 90%) for donor cells from H65-only vaccinated mice (Fig. 4D). In contrast, we found no significant differences in the percentage of H74-specific CD4 T cells in the parenchyma regardless of whether the cells came from H74 only or BCG-H74–vaccinated animals (range 78.0–99.4 and 72.0–99.4%, respectively, with means of 85.0 and 84.2%). These results clearly demonstrate that pre-existing BCG immunity significantly impacts the functionality of the T cells induced by subunit booster vaccines and that this mechanism can be efficiently bypassed by designing vaccines that selectively incorporate BCG-complementing TB Ags. Discussion The capacity of CD4 T cells to protect against M. tuberculosis is governed by their differentiation state and ability to localize to the site of infection (37). M. bovis BCG vaccination primes a polyfunctional CD4 T cell response that differentiates over time and gradually loses the ability to produce IL-2, proliferate, and to localize to the site of infection, which results in a loss of long-term protective efficacy (11–13). Heterologous prime–boost strategies, in which BCG vaccination is followed by a subunit vaccine boost, are aiming at improving the BCG-induced adaptive immunity in terms of magnitude, durability, and quality of the CD4 T cell response (38). In this study, we tested whether pre-existing BCG immunity impacts the vaccine response of either a “traditional” BCG booster vaccine (H65) or a complementary vaccine based on ESX-1–associated Ags that are specific for M. tuberculosis (H74 and H64, referred to collectively as “ESX-1 vaccines”). In the initial characterization of the ESX-1 vaccines, we demonstrated significant long-term protection in M. tuberculosis H37Rv–challenged guinea pigs as well as M. tuberculosis Erdman–challenged mice. To extend the study beyond the conventional laboratory M. tuberculosis strains, we challenged vaccinated mice with four different strains of M. tuberculosis belonging to three phylogenetically different lineages. The ESX-1 vaccine induced significant protection against all four strains, suggesting that the vaccine will be effective against a broad range of clinical isolates. Two of the selected isolates were part of the W-Beijing family of M. tuberculosis strains, one of them being the hypervirulent M. tuberculosis Beijing HN878. The Beijing strains are particularly relevant to include in this screening as they are highly prevalent, overrepresented among drug-resistant isolates (39), and significantly associated with HIV coinfection in human cases of TB meningitis (40). In a conventional protection readout at 4 wk postinfection, both the ESX-1 vaccine and BCG were highly protective against M. tuberculosis Beijing HN878. This was also true at the later time point for the ESX-1 vaccine, but in line with earlier studies (27, 41), BCG’s protective efficacy waned at the later stage of infection. In the prime–boost model, our data indicates that BCG vaccination has a major influence on the immune responses induced by subsequent subunit vaccines, depending on whether the Ags are shared with BCG or not. Specifically, we observed that boosting BCG with H65 only marginally changed the specific CD4 T cell differentiation status with little or no improvement of the protection. In contrast, BCG priming had minimal influence on the differentiation and functionality of CD4 T cells induced by the ESX-1 vaccine (not sharing Ags with BCG). The ESX-1 vaccine–specific T cells retained their differentiation status after M. tuberculosis infection, and their ability to enter the infected lung parenchyma was increased compared with the T cells in the H65-boosted group. As a result, the ESX-1 vaccine significantly augmented an already strong BCG-induced protection, and we obtained 1.8–2.9 log10 reduction in the lung bacterial load. Importantly, the BCG-priming vaccine was administered more than 6 mo prior to subunit vaccination, suggesting that it was the BCG “T cell imprint,” rather than ongoing bacterial multiplication, that influenced the response of the subunit booster vaccine. In other words, the BCG-specific CD4 T cells appeared to be “locked” with minimal capacity for reprograming into potentially more favorable phenotypes 6 mo later. This is likely because highly differentiated Th1 cells exhibit limited functional plasticity (42, 43) and that subunit boosting merely expands the existing pool (or a subset) of BCG-imprinted CD4 T cells. In addition, BCG may also induce a specific regulatory T cell response that could influence booster vaccines, although this was not investigated in this study (44, 45). Regardless, de novo priming of CD4 T cells using ESX-1–associated (or other M. tuberculosis–specific) Ags bypass this issue, which could be a useful strategy to increase durable protection in BCG-vaccinated populations. A limitation of this study was a lack of specific homing-related makers in the analysis, and in future studies, it will be important to establish whether vaccine-induced reduction of T cell differentiation leads to increased expression of such markers and/or increased contact between Ag-specific CD4 T cells and M. tuberculosis–infected macrophages in the lung. The mouse model has been extensively used to evaluate new prime–boost strategies using M. bovis BCG and subunit vaccines. The results have varied from no additional protection to almost 2 log10 protection compared with the BCG control group (46–48). It is difficult to do a comparative evaluation of these results as the studies differ with regard to vaccine design, mouse strain, M. tuberculosis challenge strain, BCG strain, and dose as well as the interval between prime–boost, boost–challenge, and challenge–sacrifice. However, by evaluating eight different available studies with BCG booster vaccines, we observed that the added protection of BCG boosting only was significant when the BCG-induced protection in itself was low (<0.5 log10) (47, 49–55). In light of our results, one explanation for this could be that a poor BCG vaccine take (or a waning response) will open up for better priming of less differentiated T cell response by the subunit vaccine. In humans, BCG will in most cases be administered to infants, and future booster vaccines are intended to be administered 10–15 y later and preferably before the protective efficacy of BCG wanes. It is not clear how pre-existing BCG immunity will influence subunit vaccination in this setting, and exposure to M. tuberculosis is also likely to play a dominant role in high-endemic areas. In this regard, results demonstrating that subunit vaccines can build on pre-existing M. tuberculosis immunity have been obtained in the recent phase IIb trial, in which M72/AS01e induced 49.7% vaccine efficacy against pulmonary disease in quantiferon positive individuals after 3 y of follow-up (56). Encouragingly, in this study, we also demonstrate that H74 vaccination significantly reduced bacterial burden in the modified Cornell model of postexposure vaccination. Similarly, regarding BCG-vaccinated quantiferon negative individuals, a recent study showed that it is possible to boost BCG protection with a subunit vaccine in adolescents and adults to some extent (57). In this study, H4:IC31 boosting led to 30.1% vaccine efficacy against sustained quantiferon conversion, and based on our data, we speculate that subunit vaccines with ESX-1–associated Ags have the potential to further improve on this result. Additionally, in the study by Nemes et al., BCG revaccination showed a vaccine efficacy of 40.5%, which has sparked renewed interest in using BCG revaccination as a readily applicable intervention (58). In such settings, the influence of recent “BCG imprinting” is likely to be significantly higher if BCG revaccination is to be combined with future subunit vaccines. In conclusion, mycobacterial priming by BCG vaccination induces highly differentiated CD4 T cells that, for at least 6 mo in the mouse model, restrict subsequent booster vaccines in priming additional protective T cells with sufficient memory and lung-homing potential. This phenomenon can efficiently be bypassed by designing vaccines with M. tuberculosis–specific Ags, like the ESX-1–associated Ags studied in this report. We suggest that future studies explore these findings in the human setting, in which it could also be investigated whether exposure to non-tuberculousis mycobacteria play a role in maintaining BCG immunity/T cell “imprint.” Disclosures C.A., N.P.H.K., I.S., E.H.K., T.L., E.M.A., M.R., I.R., P.A., and R.M. are employed by Statens Serum Institut, a nonprofit government research facility of which H56, H64, and H74 and the CAF01 adjuvant are proprietary products. C.A., P.A., and R.M. are coinventors of patents covering ESX-1–based vaccines. Acknowledgments We thank Joshua Woodworth for input on data interpretation and gratefully acknowledge Vivi Andersen and Camilla Rasmussen at Statens Serum Institut for excellent technical assistance. Footnotes This work was supported by The Danish Research Council (DFF - 7016-00310), the National Institutes of Health/National Institute of Allergy and Infectious Diseases (Grant 1R01AI135721-01), the European Union’s Horizon 2020 Framework Programme for Research and Innovation under Grant Agreement 643381 as part of the TBVAC2020 Consortium, and the National Institutes of Health/National Institute of Allergy and Infectious Diseases program Advanced Small Animal Models for the Testing of Candidate Therapeutic and Preventative Interventions against Mycobacteria (HHSN272201000009I-003, Task Order 12) at Colorado State University. The online version of this article contains supplemental material. Abbreviations used in this article: BCG Bacillus Calmette–Guérin FDS functional differentiation score MIRU mycobacterial interspersed repetitive unit TB tuberculosis. Received May 18, 2020. Accepted August 5, 2020. Copyright © 2020 by The American Association of Immunologists, Inc. This article is distributed under The American Association of Immunologists, Inc., Reuse Terms and Conditions for Author Choice articles. References ↵WHO. 2018. Global Tuberculosis Report 2018. World Health Organization, Geneva, Switzerland. ↵Trunz, B. B., P. Fine, C. Dye. 2006. Effect of BCG vaccination on childhood tuberculous meningitis and miliary tuberculosis worldwide: a meta-analysis and assessment of cost-effectiveness. Lancet 367: 1173–1180.OpenUrlCrossRefPubMed ↵Mangtani, P., I. Abubakar, C. Ariti, R. Beynon, L. Pimpin, P. E. M. Fine, L. C. Rodrigues, P. G. Smith, M. Lipman, P. F. Whiting, J. A. Sterne. 2014. Protection by BCG vaccine against tuberculosis: a systematic review of randomized controlled trials. Clin. Infect. Dis. 58: 470–480.OpenUrlCrossRefPubMed ↵Leveton, C., S. Barnass, B. Champion, S. Lucas, B. De Souza, M. Nicol, D. Banerjee, G. Rook. 1989. T-cell-mediated protection of mice against virulent Mycobacterium tuberculosis. Infect. Immun. 57: 390–395. ↵Green, A. M., R. Difazio, J. L. Flynn. 2013. IFN-γ from CD4 T cells is essential for host survival and enhances CD8 T cell function during Mycobacterium tuberculosis infection. J. Immunol. 190: 270–277. ↵Moguche, A. O., M. Musvosvi, A. Penn-Nicholson, C. R. Plumlee, H. Mearns, H. Geldenhuys, E. Smit, D. Abrahams, V. Rozot, O. Dintwe, et al. 2017. Antigen availability shapes T cell differentiation and function during tuberculosis. Cell Host Microbe 21: 695–706.e5.OpenUrlCrossRefPubMed ↵Sallin, M. A., S. Sakai, K. D. Kauffman, H. A. Young, J. Zhu, D. L. Barber. 2017. Th1 differentiation drives the accumulation of intravascular, non-protective CD4 T cells during tuberculosis. Cell Rep. 18: 3091–3104.OpenUrlCrossRef ↵He, H., P. N. Nehete, B. Nehete, E. Wieder, G. Yang, S. Buchl, K. J. Sastry. 2011. Functional impairment of central memory CD4 T cells is a potential early prognostic marker for changing viral load in SHIV-infected rhesus macaques. PLoS One 6: e19607. ↵Sakai, S., K. D. Kauffman, J. M. Schenkel, C. C. McBerry, K. D. Mayer-Barber, D. Masopust, D. L. Barber. 2014. Cutting edge: control of Mycobacterium tuberculosis infection by a subset of lung parenchyma-homing CD4 T cells. J. Immunol. 192: 2965–2969. ↵Srivastava, S., J. D. Ernst. 2013. Cutting edge: direct recognition of infected cells by CD4 T cells is required for control of intracellular Mycobacterium tuberculosis in vivo. J. Immunol. 191: 1016–1020. ↵Lindenstrøm, T., A. Moguche, M. Damborg, E. M. Agger, K. Urdahl, P. Andersen. 2018. T cells primed by live mycobacteria versus a tuberculosis subunit vaccine exhibit distinct functional properties. EBioMedicine 27: 27–39.OpenUrl Perdomo, C., U. Zedler, A. A. Kühl, L. Lozza, P. Saikali, L. E. Sander, A. Vogelzang, S. H. E. Kaufmann, A. Kupz. 2016. Mucosal BCG vaccination induces protective lung-resident memory T cell populations against tuberculosis. mBio 7: e01686-16. ↵Orme, I. M. 2010. The Achilles heel of BCG. Tuberculosis (Edinb.) 90: 329–332.OpenUrlCrossRef ↵Rodo, M. J., V. Rozot, E. Nemes, O. Dintwe, M. Hatherill, F. Little, T. J. Scriba. 2019. A comparison of antigen-specific T cell responses induced by six novel tuberculosis vaccine candidates. PLoS Pathog. 15: e1007643. ↵Woodworth, J. S., S. B. Cohen, A. O. Moguche, C. R. Plumlee, E. M. Agger, K. B. Urdahl, P. Andersen. 2017. Subunit vaccine H56/CAF01 induces a population of circulating CD4 T cells that traffic into the Mycobacterium tuberculosis-infected lung. Mucosal Immunol. 10: 555–564.OpenUrlCrossRefPubMed ↵Hoang, T., C. Aagaard, J. Dietrich, J. P. Cassidy, G. Dolganov, G. K. Schoolnik, C. V. Lundberg, E. M. Agger, P. Andersen. 2013. ESAT-6 (EsxA) and TB10.4 (EsxH) based vaccines for pre- and post-exposure tuberculosis vaccination. PLoS One 8: e80579. Billeskov, R., T. Lindenstrøm, J. Woodworth, C. Vilaplana, P. J. Cardona, J. P. Cassidy, R. Mortensen, E. M. Agger, P. Andersen. 2018. High antigen dose is detrimental to post-exposure vaccine protection against tuberculosis. Front. Immunol. 8: 1973.OpenUrl Henao-Tamayo, M., G. S. Palaniswamy, E. E. Smith, C. A. Shanley, B. Wang, I. M. Orme, R. J. Basaraba, N. M. DuTeau, D. Ordway. 2009. Post-exposure vaccination against Mycobacterium tuberculosis. Tuberculosis (Edinb.) 89: 142–148.OpenUrl Turner, J., E. R. Rhoades, M. Keen, J. T. Belisle, A. A. Frank, I. M. Orme. 2000. Effective preexposure tuberculosis vaccines fail to protect when they are given in an immunotherapeutic mode. Infect. Immun. 68: 1706–1709. ↵Taylor, J. L., O. C. Turner, R. J. Basaraba, J. T. Belisle, K. Huygen, I. M. Orme. 2003. Pulmonary necrosis resulting from DNA vaccination against tuberculosis. Infect. Immun. 71: 2192–2198. ↵Knudsen, N. P., S. Nørskov-Lauritsen, G. M. Dolganov, G. K. Schoolnik, T. Lindenstrøm, P. Andersen, E. M. Agger, C. Aagaard. 2014. Tuberculosis vaccine with high predicted population coverage and compatibility with modern diagnostics. Proc. Natl. Acad. Sci. USA 111: 1096–1101. ↵Aguilo, N., J. Gonzalo-Asensio, S. Alvarez-Arguedas, D. Marinova, A. B. Gomez, S. Uranga, R. Spallek, M. Singh, R. Audran, F. Spertini, C. Martin. 2017. Reactogenicity to major tuberculosis antigens absent in BCG is linked to improved protection against Mycobacterium tuberculosis. Nat. Commun. 8: 16085.OpenUrlCrossRef ↵Pym, A. S., P. Brodin, L. Majlessi, R. Brosch, C. Demangel, A. Williams, K. E. Griffiths, G. Marchal, C. Leclerc, S. T. Cole. 2003. Recombinant BCG exporting ESAT-6 confers enhanced protection against tuberculosis. Nat. Med. 9: 533–539.OpenUrlCrossRefPubMed Gröschel, M. I., F. Sayes, S. J. Shin, W. Frigui, A. Pawlik, M. Orgeur, R. Canetti, N. Honoré, R. Simeone, T. S. van der Werf, et al. 2017. Recombinant BCG expressing ESX-1 of Mycobacterium marinum combines low virulence with cytosolic immune signaling and improved TB protection. Cell Rep. 18: 2752–2765.OpenUrlCrossRef ↵Bottai, D., W. Frigui, S. Clark, E. Rayner, A. Zelmer, N. Andreu, M. I. de Jonge, G. J. Bancroft, A. Williams, P. Brodin, R. Brosch. 2015. Increased protective efficacy of recombinant BCG strains expressing virulence-neutral proteins of the ESX-1 secretion system. Vaccine 33: 2710–2718.OpenUrlCrossRefPubMed ↵Aagaard, C. S., T. T. Hoang, C. Vingsbo-Lundberg, J. Dietrich, P. Andersen. 2009. Quality and vaccine efficacy of CD4+ T cell responses directed to dominant and subdominant epitopes in ESAT-6 from Mycobacterium tuberculosis. J. Immunol. 183: 2659–2668. ↵Aagaard, C., T. Hoang, J. Dietrich, P. J. Cardona, A. Izzo, G. Dolganov, G. K. Schoolnik, J. P. Cassidy, R. Billeskov, P. Andersen. 2011. A multistage tuberculosis vaccine that confers efficient protection before and after exposure. Nat. Med. 17: 189–194.OpenUrlCrossRefPubMed ↵Cole, S. T., R. Brosch, J. Parkhill, T. Garnier, C. Churcher, D. Harris, S. V. Gordon, K. Eiglmeier, S. Gas, C. E. Barry III., et al. 1998. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393: 537–544.OpenUrlCrossRefPubMed ↵Behr, M. A., M. A. Wilson, W. P. Gill, H. Salamon, G. K. Schoolnik, S. Rane, P. M. Small. 1999. Comparative genomics of BCG vaccines by whole-genome DNA microarray. Science 284: 1520–1523. ↵Millington, K. A., S. M. Fortune, J. Low, A. Garces, S. M. Hingley-Wilson, M. Wickremasinghe, O. M. Kon, A. Lalvani. 2011. Rv3615c is a highly immunodominant RD1 (region of difference 1)-dependent secreted antigen specific for Mycobacterium tuberculosis infection. Proc. Natl. Acad. Sci. USA 108: 5730–5735. ↵van Pinxteren, L. A., P. Ravn, E. M. Agger, J. Pollock, P. Andersen. 2000. Diagnosis of tuberculosis based on the two specific antigens ESAT-6 and CFP10. Clin. Diagn. Lab. Immunol. 7: 155–160.OpenUrlCrossRefPubMed ↵Coscolla, M., S. Gagneux. 2010. Does M. tuberculosis genomic diversity explain disease diversity? Drug Discov. Today Dis. Mech. 7: e43–e59.OpenUrlCrossRefPubMed ↵Manca, C., L. Tsenova, S. Freeman, A. K. Barczak, M. Tovey, P. J. Murray, C. Barry, G. Kaplan. 2005. Hypervirulent M. tuberculosis W/Beijing strains upregulate type I IFNs and increase expression of negative regulators of the Jak-Stat pathway. J. Interferon Cytokine Res. 25: 694–701.OpenUrlCrossRefPubMed ↵Nandakumar, S., S. Kannanganat, J. E. Posey, R. R. Amara, S. B. Sable. 2014. Attrition of T-cell functions and simultaneous upregulation of inhibitory markers correspond with the waning of BCG-induced protection against tuberculosis in mice. PLoS One 9: e113951. ↵Seder, R. A., P. A. Darrah, M. Roederer. 2008. T-cell quality in memory and protection: implications for vaccine design. [Published erratum appears in 2008 Nat. Rev. Immunol. 8: 486.] Nat. Rev. Immunol. 8: 247–258.OpenUrlCrossRefPubMed ↵McCune, R. M. Jr.., W. McDermott, R. Tompsett. 1956. The fate of Mycobacterium tuberculosis in mouse tissues as determined by the microbial enumeration technique. II. The conversion of tuberculous infection to the latent state by the administration of pyrazinamide and a companion drug. J. Exp. Med. 104: 763–802.OpenUrlAbstract ↵Sakai, S., K. D. Mayer-Barber, D. L. Barber. 2014. Defining features of protective CD4 T cell responses to Mycobacterium tuberculosis. Curr. Opin. Immunol. 29: 137–142.OpenUrlCrossRefPubMed ↵Lewinsohn, D. A., D. M. Lewinsohn, T. J. Scriba. 2017. Polyfunctional CD4+ T cells as targets for tuberculosis vaccination. Front. Immunol. 8: 1262.OpenUrlCrossRefPubMed ↵Glynn, J. R., J. Whiteley, P. J. Bifani, K. Kremer, D. van Soolingen. 2002. Worldwide occurrence of Beijing/W strains of Mycobacterium tuberculosis: a systematic review. Emerg. Infect. Dis. 8: 843–849.OpenUrlCrossRefPubMed ↵Caws, M., G. Thwaites, K. Stepniewska, T. N. Nguyen, T. H. Nguyen, T. P. Nguyen, N. T. Mai, M. D. Phan, H. L. Tran, T. H. Tran, et al. 2006. Beijing genotype of Mycobacterium tuberculosis is significantly associated with human immunodeficiency virus infection and multidrug resistance in cases of tube
|
|
Scooped by
Gilbert C FAURE
January 4, 2020 1:32 PM
|
Mycobacterium tuberculosis (M.tb) is responsible for more deaths globally than any other pathogen. The only available vaccine, bacillus Calmette-Guérin (BCG), has variable efficacy throughout the world. A more effective vaccine is urgently needed. The immune response against tuberculosis relies, at least in part, on CD4+ T cells. Protective vaccines require the induction of antigen-specific CD4+ T cells via mycobacterial peptides presented by MHC class-II in infected macrophages. In order to identify mycobacterial antigens bound to MHC, we have immunoprecipitated MHC class-I and class-II complexes from THP-1 macrophages infected with BCG, purified MHC class-I and MHC class-II peptides and analysed them by liquid chromatography tandem mass spectrometry. We have successfully identified 94 mycobacterial peptides presented by MHC-II and 43 presented by MHC-I, from 76 and 41 antigens, respectively. These antigens were found to be highly expressed in infected macrophages. Gene ontology analysis suggests most of these antigens are associated with membranes and involved in lipid biosynthesis and transport. The sequences of selected peptides were confirmed by spectral match validation and immunogenicity evaluated by IFN-gamma ELISpot against peripheral blood mononuclear cell from volunteers vaccinated with BCG, M.tb latently infected subjects or patients with tuberculosis disease. Three antigens were expressed in viral vectors, and evaluated as vaccine candidates alone or in combination in a murine aerosol M.tb challenge model. When delivered in combination, the three candidate vaccines conferred significant protection in the lungs and spleen compared with BCG alone, demonstrating proof-of-concept for this unbiased approach to identifying new candidate antigens. Protective vaccines against Mycobacterium tuberculosis (M.tb), such as bacillus Calmette-Guérin (BCG), trigger strong CD4 T-cell responses specific to mycobacterium peptides, but their efficacy is variable. Paulo Bettencourt and colleagues now identify a set of mycobacterium peptides presented by BCG-infected macrophages via major compatibility complexes (MHC), and show that three of these antigens can be combined to formulate a vaccine that confers improved protection to Mtb infection in mice. After identifying 94 MHC-II-associated and 43 MHC-I-associated mycobacterium peptides, the researchers performed immunogenicity assays with peripheral blood mononuclear cells from BCG-vaccinated donors, latent Mtb-infected patients and patients with tuberculosis, and show that a set of these peptides was recognised by the immune cells, validating their potential as possible components for new Mtb vaccine formulations. These findings further support the value of immunopeptidomics for the identification of new antigens for effective vaccine alternatives.
|
|
Scooped by
Krishan Maggon
August 29, 2019 10:16 AM
|
Tuberculosis (TB) vaccine research has reached a unique point in time. Breakthrough findings in both the basic immunology of Mycobacterium tuberculosis infection and the clinical development of TB vaccines suggest, for the first time since the discovery of the Mycobacterium bovis bacillus Calmette–Guérin (BCG) vaccine more than a century ago, that a novel, efficacious TB vaccine is imminent. Here, we review recent data in the light of our current understanding of the immunology of TB infection and discuss the identification of biomarkers for vaccine efficacy and the next steps in the quest for an efficacious vaccine that can control the global TB epidemic.
|
|
Scooped by
Krishan Maggon
September 24, 2018 9:17 AM
|
Summary Interleukin‐17 (IL‐17) is a pro‐inflammatory cytokine and is involved in the development of many diseases. Recent studies have revealed that IL‐17‐producing γδ T cells (γδ17 cells) in addition to IL‐17‐producing CD4+ T cells [T helper type 17 (Th17) cells] are often the main producers of IL‐17 in mouse models of inflammatory diseases. γδ T cells are functionally committed during intra‐thymic differentiation. γδ thymocytes capable of producing IL‐17, which express the transcription factor retinoic‐acid‐receptor‐related orphan receptor γt and the signature cytokine receptor IL‐23R, leave the thymus, and produce IL‐17 rapidly by the stimulation with IL‐1β and IL‐23 in the periphery. Therefore, γδ17 cells play important roles in the early phase of host defence against pathogens and in inflammatory diseases. γδ T cells that can produce IL‐17 are also increased in the skin of patients with psoriasis and in peripheral blood of patients with ankylosing sclerosis. Indeed, the therapy targeting IL‐17 has been approved or is in clinical trials, and proved to be very efficient to treat psoriasis, psoriatic arthritis and ankylosing sclerosis. In this review, we discuss recent knowledge about the pathophysiological function of γδ17 cells in infection and inflammatory diseases and therapeutic advances targeting IL‐17. Introduction Interleukin‐17A (IL‐17A, called ‘IL‐17’ hereafter) is a member of the IL‐17 family.1 The binding of IL‐17 to the heterodimeric receptor consisting of IL‐17RA and IL‐17RC subunits transduces signals to activate a group of cytokines and chemokines such as tumour necrosis factor, IL‐1, IL‐6, granulocyte colony‐stimulating factor, CXCL1 and CXCL2 through activation of the actin related gene 1‐TNF receptor associated factor 6–nuclear factor‐κB axis in the downstream.1 The function of IL‐17 is pleiotropic. It plays a crucial role in the host defence against bacterial and fungal infection by inducing pro‐inflammatory cytokines and chemokines, recruiting neutrophils, and activating T cells and B cells.1, 2 Mice deficient for IL‐17RA are highly susceptible to Klebsiella pneumoniae3 and IL‐17‐deficient mice are susceptible to bacterial and fungal infection.4, 5 Interleukin‐17 is also implicated in various inflammatory/autoimmune disease models such as experimental autoimmune encephalomyelitis (EAE), arthritis in IL‐1 receptor antagonist‐deficient (Il1rn–/–) mice and imiquimod‐induced psoriatic dermatitis in mice.6-10 Anti‐IL‐17 and anti‐IL‐17RA antibodies are effective to treat patients with psoriasis and psoriatic arthritis.11-13 Interleukin‐17 is also important for the maintenance of intestinal barrier integrity and its functional deficiency causes the development of inflammatory bowel diseases.14-16 In contrast, suppression of IL‐17F, another highly homologous member of IL‐17 family, is suggested to be beneficial for the treatment of inflammatory bowel diseases.17 Interleukin‐17 was initially found to be produced by helper CD4+ T [T helper type 17 (Th17)] cells, but subsequent studies showed that innate immune cells and innate‐like immune cells are also important sources of IL‐17 in inflamed tissues.18, 19 γδ T cells are the principal source of IL‐17 in some mouse inflammatory disease models and thereby exert considerable impact on disease development and progression.20-24 The IL‐17‐producing γδ T cells (γδ17 cells) share many features with Th17 cells, such as cell surface expression of IL‐23R and CCR6 and the expression of transcriptional factor retinoic‐acid‐receptor‐related orphan receptor γt (RORγt). To induce IL‐17, naive T cells have to differentiate into Th17 cells in the periphery by the stimulation with T‐cell receptor (TCR) and cytokines such as IL‐6 and transforming growth factor‐β. In contrast, the functional potential to produce IL‐17 in γδ17 cells is already established during intra‐thymic development25-27 and IL‐17 is directly induced by IL‐23 and IL‐1 without TCR stimulation in the periphery. This pre‐programming contributes to rapid IL‐17 production in peripheral tissues in the early phase of pathogen infection. In this review, we would like to introduce the roles of γδ17 cells in inflammatory diseases and recent therapeutic advances targeting IL‐17. γδ T‐cell subsets and their development γδ T cells and αβ T cells are generated in thymus from common progenitor cells. Unlike αβ T cells, γδ T cells are functionally committed during intra‐thymic differentiation.28, 29 In mice, the TCR‐γ locus consists of seven Vγ (Vγ1–Vγ7) genes (Heilig & Tonegawa's nomenclature30) that are closely correlated with the effector function, although Vγ3 is a pseudogene in most mouse strains.31 Production of IL‐17 is mostly limited to Vγ4+ and Vγ6+ γδ T cells,32 although Vγ1+ γδ T cells also produce IL‐17 in some cases.33 On the other hand, interferon‐γ (IFN‐γ) production is associated with Vγ1+, Vγ5+ and Vγ7+ γδ T cells. Although overall gene expression patterns are similar between Vγ4+ and Vγ6+ subsets,34 each subset has distinct features (Fig. 1). Vγ6+ γδ T cells express the invariant Vγ6/Vδ1 TCR, develop only in the late embryonic thymus and preferentially localize to the uterus, vagina, lung, dermis and peritoneal cavity.35, 36 On the other hand, Vγ4+ γδ T cells develop in both fetal and adult thymus, have more diverse TCR repertoire and reside in the dermis, lung, liver and secondary lymphoid organs.37, 38 In addition to RORγt, transcription factors such as Blk,39 Hes‐1,40 nuclear factor‐κB,41 Sox4 and Sox1342 are also important for γδ17 cell development. Transforming growth factor‐β and IL‐7 are required for γδ17 thymocyte development and expansion, respectively.43-45 Epigenetic and transcriptional regulation during γδ17 cell differentiation has been reviewed elsewhere.46 γδ thymocytes capable of producing IL‐17, which express the transcription factor RORγt and the signature cytokine receptor IL‐23R,34 leave the thymus as functionally committed cells,47 and produce IL‐17 directly by the stimulation with IL‐1β and IL‐23 in the periphery. Although IL‐23R is constitutively expressed on γδ17 cells, the expression of IL‐1R in peripheral γδ17 cells is tissue‐dependent.48 In addition to IL‐1R and IL‐23R, the expression of scavenger receptor 2 (Scart 2)49 and CCR6,27 and the lack of CD12225 and CD27 expression26 are often used as markers for γδ17 cells, with the exception for IL‐17‐producing Vγ1+ γδ T cells.33 These phenotypes, established during thymic development, distinguish γδ17 cells from IFN‐γ‐producing γδ T (γδIFN‐γ) cells (Fig. 2). γδ17 cells that develop before birth persist in adult mice as self‐renewing, long‐lived cells.50 The requirement of TCR signalling for γδ17 cell development is not fully understood.51 Early T‐cell precursors can produce IL‐17 before TCR recombination50 and Sox4 and Sox13 are expressed before activation with TCR signalling.42 Antigen‐naive γδ T cells in the thymus differentiate into IL‐17 producers, whereas antigen‐experienced cells make IFN‐γ.28 Furthermore, Vγ5+Vδ1+ thymocytes induce Egr3 upon recognition of Skint‐1 expressed on thymic epithelial cells, resulting in the induction of IFN‐γ expression and suppression of RORγt and Sox13 expression.52 Thus, TCR signalling seems to direct γδ thymocytes to differentiate into IFN‐γ‐producing γδ T cells by suppressing the ‘default’ IL‐17 programme. The mechanism of IL‐17 production in γδ17 cells γδ17 cells are functionally committed in the thymus, producing IL‐17 in the periphery after stimulation with IL‐1β and IL‐23 without additional TCR stimulation.21, 24 The ‘ready‐to‐go’ phenotype of γδ17 cells is especially efficient for early‐stage pathogen clearance. A combination of IL‐1β and IL‐23, but not IL‐1β or IL‐23 alone, is required to induce IL‐17 by γδ17 cells.20 Further study revealed that IL‐23 is required for the induction of IL‐1R, and IL‐1β is essential for the induction of IL‐17.20 However, because IL‐1β alone does not induce IL‐17 production in Il1rn−/− mouse‐derived splenic γδ T cells20 and in normal peritoneum‐ and lung‐derived γδ T cells, in which high levels of IL‐1R are expressed,48 IL‐23 may play other roles than up‐regulating IL‐1R in the induction of IL‐17 expression in γδ T cells. In this context, RORγt expression is up‐regulated by IL‐1β and IL‐23 in a synergistic manner.20 The inflammatory cytokine IL‐18,53 complement C5a,54 the ligand of Toll‐like receptors 1 and 2, and dectin‐155 also induce IL‐17 in collaboration with IL‐23. Although γδ17 cells typically behave as innate‐like immune cells, IL‐17 induction by TCR signalling is also reported. Mouse and human γδ T cells recognize an algal protein, phycoerythrin, and differentiate to IL‐17‐producing cells after immunization by this antigen.56 These studies, in combination with cell reconstitution studies,50 suggest that ‘natural’ γδ17 cells (Vγ6+ and part of Vγ4+) acquire IL‐17‐producing ability in the fetal thymus and do not require TCR stimulation in the periphery, whereas ‘inducible’ γδ17 cells (mostly Vγ4+) that develop after birth produce IL‐17 upon encounter with antigens57 (Fig. 1). Notably, phycoerythrin antigen stimulation induces IL‐1R expression on γδ T cells.56 Therefore, γδ TCR activation may make ‘inducible’ γδ17 cells respond to IL‐1β and IL‐23 to induce IL‐17. A similar activation mechanism is also suggested in Th17 cell differentiation; IL‐1R expression is increased upon Th17 differentiation from naive CD4+ T cells,58 and the polarized Th17 cells can produce IL‐17 by IL‐1 and IL‐23 in the absence of TCR stimulation.59 However, the molecular basis for the ‘inducible’ state and the difference between ‘naive’ and ‘inducible’ γδ17 cells remain to be elucidated. The pathogenic roles of γδ17 cells in mouse inflammatory disease models Il17–/– mice show significantly reduced severity in various inflammatory and autoimmune disease models, such as collagen‐induced arthritis,6 Il1rn−/− mouse arthritis,7 EAE8 and imiquimod (IMQ)‐induced skin inflammation,9, 10 suggesting critical roles of IL‐17 in inflammatory/autoimmune diseases. γδ17 cells are detected in inflamed tissues of these disease models. However, the roles of γδ T cells in the development of diseases and the responsible γδ subset are different in different models (Table 1). As no conditional Il17–/– mice in which the Il17 gene is deleted specifically in γδ17 cells are available, the pathogenicity of γδ17 cells has been analyzed using γδ T‐cell‐deficient mice (Tcrd‐/‐ mice) or the γδ17 cell subset from these disease models. Disease Disease model Dominant subset Induction of γδ17 cells γδ17 function References Rheumatoid arthritis (RA) collagen‐induced arthritis Vγ4+ Vδ4+ Mycobacterium tuberculosis components in complete Freund's adjuvant (CFA) or subsequent cytokine induction Promote Th17 cells 22, 24, 60 Il1rn−/− mice Vγ6+ Vδ1+ Up‐regulation of interleuki ‐1 receptor (IL‐1R) IL‐17‐induced hyperinflammation 20 Spondyloarthritides (SpA) IL‐23 overexpression Vγ6+ Hyper IL‐23 induction IL‐17‐induced hyperinflammation 65 Multiple sclerosis (MS) EAE Vγ4+ IL‐1β and IL‐23 from dendritic cells (DC) induced by Mycobacterium tuberculosis components in CFA Promote Th17 cells, and suppress regulatory T cells 21, 61 Uveitis EAU Vγ4+ Vδ4+ Components in CFA or subsequent cytokine induction Promote Th17 cells 104 Psoriasis IMQ Vγ4+ and Vγ6+ Imiquimod (IMQ)‐induced IL‐23 from DC IL‐17 production and subsequent neutrophil inflammation 9, 10, 23, 35, 42, 64 IL‐23 Hyper IL‐23 induction IL‐17 production and subsequent neutrophil inflammation 23 Uveoretinitis in autoimmune polyendocrinopathy–candidiasis–ectodermal dystrophy (APCED) Aire−/− mice Vγ6+ Vδ1+ Up‐regulation of IL‐7 in the thymus IL‐17‐induced hyperinflammation 44 Skin graft rejection Male to female skin transplantation Vγ4+ Accumulation CCR6+ γδ17 cells in skin graft IL‐17 promotes the accumulation of DC in draining lymph nodes to subsequently activate Th17 cells 66 In the collagen‐induced arthritis model, both γδ17 cells and Th17 cells are found in joints and draining lymph nodes and the majority of the γδ17 cells are Vγ4+/Vδ4+.22, 24 Vγ4+ cell depletion reduces Th17 cell number60 as well as arthritis severity,22 suggesting that the Vγ4+/Vδ4+subset aggravates disease by promoting a Th17 cell response. Some components of heat‐killed Mycobacterium tuberculosis in complete Freund's adjuvant or inflammatory cytokines induced by the adjuvant are suggested to induce Vγ4+/Vδ4+ cell expansion.24, 60 Il1rn−/− mice develop arthritis spontaneously in an IL‐17‐dependent manner.7 In these mice, however, only γδ17 cells are the IL‐17 producer in the joints, although both γδ17 cells and Th17 cells are detected in the draining lymph nodes.20 IL‐17‐GFP reporter mice reveal that the Vγ6+/Vδ1+ cells predominantly produce IL‐17 in affected joints. Adoptive transfer of Il1rn–/– T cells into scid/scid mice shows that only a mixture of γδ T and CD4+ T cells, but not γδ T cells or CD4 T cells alone, can induce arthritis. Moreover, γδ17 cells localize in joints only when γδ T cells are transferred together with CD4+ T cells. These observations suggest that CD4+ T cells are required for γδ17 cells to localize in joints, and IL‐17 from γδ17 cells drives the development of arthritis. Interestingly, Vγ6+ γδ17 cells in Il1rn−/− mice intrinsically express IL‐1R at high levels, indicating that these cells are ready for IL‐17 production. Hence, IL‐17 derived from different γδ17 cell subsets is suggested to play a crucial role in the development of arthritis in mouse models. The mouse model of multiple sclerosis (MS), EAE is another model in which IL‐17 plays a crucial role in the pathogenesis. After induction of EAE, γδ17 cells as well as Th17 cells are found in the brain, with Vγ4+ cells as the major component.21 Tcrd−/− mice delay the onset of disease and reduce the clinical scores. γδ T cells activated with IL‐1β and IL‐23 promote IL‐17 production by CD4+ T cells in vitro and co‐transfer of CD4+ and γδ T cells promote development of EAE, suggesting that γδ17 cells act in an amplification loop for IL‐17 production by Th17 cells.21 Interleukin‐23‐activated γδ T cells also prevent regulatory T‐cell function, resulting in the enhancement of αβ T‐cell responses and EAE development.61 A pathogenic role for γδ17 cells is implicated in psoriasis. Vγ5+/Vδ1+ γδ T cells, also called dendritic epidermal T cells, uniquely residing in epidermis produce IFN‐γ and participate in immunosurveillance.62 On the other hand, dermis contains Vγ4+ and Vγ6+ subsets responsible for IL‐17 production. The pathogenicity of γδ17 cells in psoriasis is well studied in a mouse model of psoriasis, IMQ‐induced dermatitis.9, 23 Imiquimod is a Toll‐like receptor‐7/8 agonist and induces IL‐17‐dependent psoriasiform dermatitis by inducing IL‐23.63 The IMQ‐induced epidermal thickening is significantly decreased in Tcrd–/– mice, but not in Tcrb–/– mice.9 Development of dermatitis is also suppressed by the deficiency of innate lymphoid cells (ILCs), suggesting that γδ17 cells and IL‐17‐producing group 3 ILCs (ILC3s) are responsible for the development of psoriasiform dermatitis in mice.9, 23 Both Vγ4+ and Vγ6+ subsets produce IL‐17 after IMQ treatment of the skin.35 Skin inflammation after IMQ treatment is significantly attenuated in Sox4−/− mice, in which dermal Vγ4+ but not dermal Vγ6+ γδ T cells are greatly reduced.42 Congenic CD45.1+ (B6.SJL) mice with naturally occurring Sox13 mutation, in which dermal Vγ4+ γδ17 cell development is defective, develop attenuated ear skin inflammation with less acanthosis and fewer epidermal neutrophil pustules upon treatment with IMQ.64 However, both wild‐type bone marrow cell‐reconstituted mice and neonatal thymocytes plus CD45.1+(B6.SJL) bone marrow cell‐reconstituted mice (in which Vγ4+ and Vγ6+ cells are predominant, respectively) similarly develop epidermal thickening with increased dermal γδ17 cells and neutrophil infiltration, suggesting that both γδ17 subsets induce IMQ‐induced dermatitis.35 Interleukin‐17F and IL‐22 from γδ T cells are also pathogenic in IMQ‐induced dermatitis.9 Interleukin‐23‐induced skin inflammation is another model of psoriasis. The IL‐17‐producing dermal cells are significantly reduced in Tcrd−/− mice accompanied with less skin inflammation and acanthosis, whereas Tcra−/− mice normally develop dermatitis,23 suggesting that γδ17 cells play major roles in the pathogenesis of psoriatic dermatitis in this model. The involvement of γδ17 cells in the development of psoriasiform dermatitis suggests that innate immune responses rather than an autoimmune reaction are important for the development of psoriatic dermatitis. The involvement of Vγ6+ γδ17 cells in the pathogenesis of uveoretinitis in Aire‐deficient mice, a model of autoimmune polyendocrinopathy–candidiasis–ectodermal dystrophy (APCED), is suggested, because Vγ6+ γδ17 thymocytes are expanded due to high levels of IL‐7 expression in Aire−/− medullary thymic epithelial cells.44 Importantly, expansion of IL‐17‐producing Vγ9+/Vδ2+ γδ T cells is observed in APCED patients, suggesting the involvement of γδ17 cells in these patients. γδ17 cells are also implicated in the pathogenesis of spondyloarthritis; the CD27− γδ T‐cell population is increased in the Achilles tendon after over‐expression of IL‐23, which induces spondyloarthritis‐like enthesitis in mice.65 The Vγ4+ subset produces IL‐17 in the skin grafts and in the host epidermis around grafts, suggesting the involvement in skin graft rejection.66 Vγ4+cell‐derived IL‐17 promotes the accumulation of mature dendritic cells in the draining lymph nodes to subsequently increase Th17 cells after skin graft transplantation.66 Migration of γδ17 cells to inflammatory sites Trafficking of γδ17 cells to the inflammatory sites is important for the development of inflammation. Naive αβ T cells express a chemokine receptor CCR7, which is important for homeostatic circulation; its expression is down‐regulated during differentiation and the inflammatory chemokine receptor CCR6 is induced on Th17 cells instead. CCR6+ Th17 cells are recruited by CCL20 to cause inflammation, as shown in SKG mice67 and the EAE model.68, 69 On the other hand, a gene array analysis shows that the expression of chemokine receptors such as CCR6, CCR2 and CXCR6 is already up‐regulated in γδ17 cells during thymic development.34 Recent studies have indicated that the CCL20–CCR6 axis is mainly required for γδ17 cell recruitment into homeostatic sites such as dermis,70 whereas the CCL2–CCR2 axis recruits γδ17 cells into inflammatory sites, including psoriatic skin,71 arthritic joints,20 central nervous system in EAE, infected mucosal tissues and tumours.70 Interestingly, γδ17 cells constitutively express both CCR2 and CCR6, but down‐regulate CCR6 expression after inflammation.70 This is consistent with the ‘ready‐to‐go’ nature of γδ17 cells and suggests a γδ17 cell‐recruiting mechanism in which γδ17 cells are released from the tissue‐specific harness to migrate into inflammatory sites by reducing the tissue‐specific homing receptor. γδ17 cells in tumours Pro‐tumour function of γδ17 cells has been demonstrated in several cancer models, including a breast cancer metastasis model72 and an ovarian cancer model,73 and the roles of γδ17 cells in the development of tumours have been reviewed elsewhere.74 Abundant γδ17 cell infiltration accompanied by immunosuppressive myeloid‐derived suppressor cells is found in human colorectal cancer with positive correlation with advanced tumour clinicopathological features, suggesting that γδ17 cells induce myeloid‐derived suppressor cell‐mediated immunosuppression.75 On the other hand, anti‐tumour function of γδ17 cells after therapeutic treatment has also been reported. γδ17 cells infiltration is observed when bladder cancer is treated by intravesical injection of Mycobacterium bovis bacillus Calmette–Guérin, (BCG) and these cells are protective against tumour development by recruiting neutrophils.76 Moreover, γδ17 cell infiltration in epithelial tumours is observed after chemotherapy and γδ17 cells enhance the recruitment of IFN‐γ‐producing CD8+ T cells that mediate the anti‐tumour function.77 γδ17 cells in pathogen clearance Interleukin‐17 plays protective roles against bacterial and fungal infection by recruiting neutrophils, activating T cells and inducing antimicrobial peptides and inflammatory cytokines.1, 4, 5 γδ17 cells produce much more IL‐17 than Th17 cells after Mycobacterium tuberculosis78 or Mycobacterium bovis BCG infection.79 Expression of IL‐17 is detected in lungs from the first day after infection with BCG and induces not only neutrophil‐mediated inflammation but also granuloma formation.79 Dermal γδ T cells also produce IL‐17 at the first day after intradermal BCG infection and induce neutrophil recruitment and antigen‐specific CD4+ T‐cell expansion.80 Rapid IL‐17 production by Vδ1+ T cells is also observed in the peritoneum after intraperitoneal infection with Escherichia coli, followed by neutrophil recruitment.81 γδ17 cells are also found in the liver after Listeria monocytogenes infection82 and in lungs after Klebsiella pneumoniae infection.83 γδ17 cells produce IL‐17 rapidly after infection with fungi, such as Candida albicans. γδ17 cells are observed in lungs after systemic C. albicans infection, and both Il17−/− and Tcrd−/− mice are defective in neutrophil recruitment and fungal clearance.84 Interleukin‐17 is produced in tongue‐resident γδ T cells as well as CD3+ CD4+ CD44hi TCR‐β+ CCR6+ natural Th17 cells within 1–2 days after oral C. albicans infection.85 As IL‐17 production in γδ T cells is found at the early stage after infection, γδ17 cells are suggested to play an important role in early host defence before establishment of acquired immunity. In humans, patients carrying mutations in either STAT3, IL‐17RA, or IL‐17F or producing anti‐IL‐17F autoantibodies are also highly susceptible to skin infection with Staphylococcus aureus and C. albicans.86, 87 However, the involvement of γδ17 cells in host defence in humans against these pathogens remains to be elucidated. Recently, pathogen‐specific memory γδ T cells have been implicated in several infection models. Memory γδ T cells are elicited in mesenteric lymph nodes after oral L. monocytogenes infection and contribute to clearance of the bacteria by promptly producing IL‐17 after secondary infection.88, 89 Interestingly, both Vγ4+ and Vγ1− Vγ4− γδ (potentially Vγ6+) T cells produce IL‐17 in the lungs as early as 2 hr after Bordetella pertussis infection, whereas the exclusively Vγ4+ subset expands in lungs 14 days after infection. Moreover, lung Vγ4+ γδ T cells produce IL‐17 in response to heat‐killed B. pertussis in the presence of antigen‐presenting cells.90 These studies suggest that Vγ4+ T cells, but not Vγ6+ T cells, behave like adaptive immunological memory cells in a pathogen‐specific manner. γδ17 cells in human inflammatory diseases Although it has been thought that psoriasis is caused by an autoimmune mechanism,91 recent studies using mouse models suggest the involvement of innate immunity.92 High frequency of γδ T cells is detected in psoriasiform dermal lesions in mice induced by IMQ, and these cells produce IL‐17 through stimulation with IL‐23,23 indicating the innate immune nature of the disease. γδ T cells, especially Vγ9+Vδ2+ cells which can produce IL‐17, are also accumulated in the skin of patients with psoriasis.93 In this report, however, Vγ9+Vδ2+ cells were activated to produce cytokines from keratinocytes by the specific antigen, suggesting that recognition of specific antigens may be important for the development of psoriasis. Because Th17 cells94 and ILC3s95 as well as γδ17 cells91 are also detected in the psoriatic skin, the involvement of autoimmunity and the main source of IL‐17 during the development of psoriasis still remain obscure in humans. Recently, several antibodies targeting IL‐17 and its receptor have been approved or are in clinical trials for the treatment of psoriasis and psoriatic arthritis.96 Secukinumab, an anti‐IL‐17 antibody, has been approved in Japan in 2014 and by the US Food and Drug Administration in 2015 for the treatment of psoriasis and psoriatic arthritis. Treatment with secukinumab for psoriasis patients in a phase III trial shows that more than half of the patients accomplish almost complete remission after 12 weeks of treatment, as determined by the Psoriasis Area and Severity Index (PASI 90), and show better efficacy than a tumour necrosis factor inhibitor (etanercept).11 Ixekizumab, another monoclonal antibody against IL‐17, and Brodalumab, a monoclonal antibody against IL‐17RA, are also effective for the treatment of psoriasis in phase III trials12, 13 and approved for the treatment of psoriasis. Secukinumab has also been approved for the treatment of ankylosing spondylitis.97 As described, the importance of IL‐17 in the development of rheumatoid arthritis (RA) is suggested in mouse models and γδ17 cells are accumulated in arthritic joints,7, 20, 22, 24 but the importance of γδ17 cells in patients with RA is controversial. A recent report shows that Vδ2+ γδ T cells accumulate in the synovium of patients with RA and produce high levels of IL‐17 as well as IFN‐γ.98 However, the predominance of IFN‐γ‐producing γδ T cells instead of γδ17 cells in affected joints is reported elsewhere.24 This discrepancy may result from the differences of medical treatment and/or stages of RA. Because the efficacy of anti‐IL‐17 treatment of RA is moderate except for some patients with specific HLA types,99, 100 γδ17 cells may not play crucial roles in the development of RA in humans. Further studies in patients with RA are necessary. Elevated IL‐17 expression in γδ T cells, but not CD4+ T cells, was found in patients with systemic juvenile idiopathic arthritis,101 and IL‐17 production was detected in CD161hi CCR6+ γδ T cells in cerebrospinal fluid of patients with MS.102 Treatment of patients with MS with secukinumab non‐significantly reduced the number of combined unique active lesions and significantly reduced the number of cumulative new gadolinium‐enhancing T1 lesions by 67%.103 Further studies are necessary to elucidate the role of γδ17 cells in the development of these diseases. Concluding remarks In this review, we discussed the development and the function of γδ17 cells and the roles of γδ17 cells in inflammatory/autoimmune diseases and host defence against pathogens. However, several important questions still remain to be elucidated. The function of TCR signalling in the thymic development of γδ17 and the functional roles of TCR signalling in the periphery upon infection and inflammation are not completely elucidated. Elucidation of epigenetic modifications of genes in ‘natural’ and ‘inducible’ γδ17 cells may provide important information. Discovery of γδ17 cell‐specific markers may provide a more efficient approach to induce γδ17 cell‐specific dysfunction in inflammatory diseases without affecting systemic Th17 cells. Most importantly, it is not clear how well mouse disease models represent the pathogenesis of human diseases, especially those of psoriasis, MS and RA. In the case of psoriasis, inhibition of IL‐17 signalling efficiently cures the symptoms both in psoriasis patients and IMQ‐induced psoriasis models, suggesting that γδ17 cells play important roles in both humans and mice. However, in MS and RA, in which the involvement of γδ17 is suggested by mouse models, the therapeutic effects of anti‐IL‐17 are not so drastic as indicated by these models, suggesting that the pathogenic mechanisms may be different in some parts between diseases in humans and mice. Clearly, further analysis of pathogenic mechanisms in patients is necessary to explain this discrepancy. Acknowledgements We thank Professor Danny Altmann, the Editor of Immunology, for the invitation to submit this review. Our laboratory is funded by Grants‐in‐Aid from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (Y.I.) and AMED (Y.I.). Disclosures The authors declare no conflict of interest. References
|
|
Scooped by
Krishan Maggon
April 2, 2018 4:45 AM
|
LepVax immunization did not exacerbate cutaneous nerve involvement due to M. leprae infection, indicating its safe use. There was no intraneural inflammation but a reduction of intra axonal edema suggested that LepVax treatment might restore some early sensory axonal function. These data indicate that post-exposure prophylaxis with LepVax not only appears safe but, unlike BCG, alleviates and delays the neurologic disruptions caused by M. leprae infection.
New data from an FDA-approved clinical trial testing the generic vaccine bacillus Calmette-Guérin to reverse advanced type 1 diabetes demonstrate a potential new mechanism by which the BCG vaccine may restore the proper immune response to the insulin-secreting islet cells of the pancreas.
Via Denis Hudrisier
|
|
Scooped by
Gilbert C FAURE
December 20, 2013 3:30 PM
|
The BCG vaccine has been found to be more effective against the most common form of tuberculosis than previously thought, according to a new study in Clinical Infectious Diseases. Bacillus Calmette Guérin (BCG) vaccine is ...
|
|
|
Scooped by
Gilbert C FAURE
November 27, 2025 3:30 AM
|
🎉 NEW PAPER PUBLISHED 🔬
Over a century after its introduction, the BCG vaccine continues to surprise us. Despite being given to billions of people, we still know remarkably little about what actually happens in the skin and blood in the hours and days right after intradermal BCG vaccination. Our team set out to change that.
In this study, conducted in Guinea-Bissau, we developed and validated a completely new experimental pipeline—bringing cutting-edge “omics” technologies into a difficult low-resource setting—to map the local (skin) and systemic (blood) immunological events following BCG.
Here are some of the most exciting methodological advances:
✨ 1. Spatial transcriptomics on 2 mm skin punch biopsies from healthy volunteers with and without a BCG scar
Using Nature’s 2020 Method of the Year, we assessed gene expression within the actual tissue architecture of the skin. For the first time, we could identify which cell types in the epidermis, dermis, and hypodermis respond to BCG, and how.
✨ 2. Liquid biopsy: Cell-free RNA (cfRNA) Profiling
By capturing cell-free RNA from plasma, we non-invasively traced molecular signals released from the skin into the bloodstream. This offers the prospect of blood-based biomarkers that reflect tissue-level vaccine responses—a major step toward scalable immune monitoring.
✨ 3. Multiomics integration
We combined spatial transcriptomics, whole-blood transcriptomics, epigenetics, proteomics, metabolomics, & advanced computer-vision analysis of dermatoscopic images.
✨ 4. Precision study in a low-resource setting
From dermatoscopic imaging (using a smartphone-based system) to standardized storage/processing workflows (see Fig. 2 in the paper), we demonstrated that highly advanced immunology is possible in West Africa—opening the door for larger trials in populations where BCG’s effects matter most.
✨ 5. Capturing early immune events never previously characterized
By sampling on day 1, 7, & 14, and stratifying participants by presence/absence of pre-existing BCG scars, this project may shed light on why BCG given at birth has such profound survival benefits—and why revaccination responses differ.
Why this matters:
Most new TB vaccines build on BCG, yet the early steps of how BCG “trains” the immune system have never been fully mapped. Understanding these local and systemic events may be important for designing better vaccines—not only against TB, but potentially against a wide range of infections where BCG has shown non-specific (heterologous) benefits.
A huge thank you to our incredible collaborators across Guinea-Bissau, Denmark and Canada. And especially to the participants in Bissau who made this study possible.
If you’re interested in immunology, systems biology, vaccine innovation or (of course) vaccine epidemiology, let me hear your thoughts and/or connect with me here on LinkedIn.
📄 The full paper is available open access (link in comments) - & we have more interesting to come from this project.

|
Suggested by
Société Francaise d'Immunologie
November 26, 2020 2:16 PM
|
Abstract In recent years, the century‐old Mycobacterium bovis Bacillus Calmette‐Guerin (BCG) vaccine against tuberculosis (TB) has been re‐evaluated for its capacity to stem the global tide of TB. ...
|
|
Suggested by
Société Francaise d'Immunologie
May 30, 2020 2:22 AM
|
To Editor COVID-19, caused by the new corona virus (SARS-CoV-2), is an emerging, rapidly evolving disease that needs rapid intervention as it shows high spread mortality rates within very short time. Interestingly, the reported cases show different severity of symptoms, ranging from mild to severe with no symptoms in some cases. Although very limited studies investigated the immune responses toward COVID-19, a recent study conducted by researchers at the Peter Doherty Institute for Infection and Immunity in Australia, assessed the immune responses in the blood from a patient with COVID-19 disease with mild severity [1]. They looked at the cellular and humoral immune responses at different time points during the infection; i.e. before, during and after resolution of the disease and recovery of the patient. Their longitudinal analysis showed a robust immune response across different cell types associates with clinical recovery. These findings are similar to what the same group have reported before in patients with influenza infection [2], [3], Accordingly, we suggest a link between the quality of the immunity and recovery from COVID-19, at least in part, in patients with mild symptoms. Indeed, different susceptibilities to COVID-19 disease were observed between different age groups where children showed lower rate of infection than adults and elderly. Although the mechanism behind these differences in infection severity and susceptibility is not clear, one possible explanation could be the difference in the quality and quantity of the immune performance that is shaped by the history of recent infections and/or vaccinations. We present here the hypothesis that the resultant immunity against prior influenza infection would, at least in part, foster immunity against SARS-CoV-2. This hypothesis is supported by which the similarity in the quality of immunity toward both viruses. and by the previous studies showing cross reactivity of immunity between Flu and coronavirus [4] due to the similarity in their structures [5], [6]. Besides the cross reactivity effect, the anti-Flu immune responses can induce bystander immunity [7] that is expected to non-specifically augment immunity against other viral infection such as SARS-CoV-2. Furthermore, influenza vaccination itself would generate sustained immunity that overall enhance immunity against SARS-CoV-2. This would explain why the rate of SARS-CoV-2 in children is low since they catch flu more than adults do [8]. As such, it is expected that their immune systems be often alarmed against influenza, generating bystander immunity that harness the immune responses against related viral infection. Under this setting, we hypothesize that children generate multifactorial immunity with the repeated influenza exposure that would offer bystander immune response in case they became infected with the new SARS-CoV-2. It might be possible also that individuals who received prior Flu vaccination might show mild severity of COVID-19 because of Flu-induced bystander effect of the generated immune responses which itself might cross react against SARS-CoV-2. Due to this cross reactivity between Flu and SARS-CoV-2, we suggest that the Flu-induced bystander immunity is more of beneficial effects to COVID-19 than those suggested by MMR and BCG vaccines [9], [10]. Indeed, the zero COVID-19 patient (the Chinese patient suspected to be the first case infected with the new corona virus) who was released from the hospital couple of weeks after her diagnosis declared that the symptoms were almost like those of her repeated flu infection [11]. Given the safety of Flu vaccine in adult, we recommend the use of Flu vaccine, at least in part, as a bystander adjuvant to minimize the severity of COVID-19 disease. We have no conflicts of interest to disclose.
|
|
Scooped by
Krishan Maggon
November 2, 2019 11:30 AM
|
Abstract Pathophysiology of graft failure (GF) occurring after allogeneic hematopoietic stem cell transplantation (HSCT) still remains elusive. We measured serum levels of several different cytokines/chemokines in 15 children experiencing GF, comparing their values with those of 15 controls who had sustained donor cell engraftment. Already at day +3 after transplantation, patients developing GF had serum levels of interferon (IFN)-γ and CXCL9 (a chemokine specifically induced by IFNγ) significantly higher than those of controls (8859±7502 vs. 0 pg/mL, P=0.03, and 1514.0±773 vs. 233.6±50.1 pg/mlL, P=0.0006, respectively). The role played by IFNγ in HSCT-related GF was further supported by the observation that a rat anti-mouse IFNγ-neutralizing monoclonal antibody promotes donor cell engraftment in Ifngr1−/−mice receiving an allograft. In comparison to controls, analysis of bone marrow-infiltrating T lymphocytes in patients experiencing GF documented a predominance of effector memory CD8+ cells, which showed markers of activation (overexpression of CD95 and downregulation of CD127) and exhaustion (CD57, CD279, CD223 and CD366). Finally, we obtained successful donor engraftment in 2 out of 3 children with primary hemophagocytic lymphohistiocytosis who, after experiencing GF, were re-transplanted from the same HLA-haploidentical donor under the compassionate use coverage of emapalumab, an anti-IFNγ monoclonal antibody recently approved by the US Food and Drug Administration for treatment of patients with primary hemophagocytic lymphohistiocytosis. Altogether, these results suggest that the IFNγ pathway plays a major role in GF occurring after HSCT. Increased serum levels of IFNγ and CXCL9 represent potential biomarkers useful for early diagnosis of GF and provide the rationale for exploring the therapeutic/preventive role of targeted neutralization of IFNγ. Introduction Graft failure (GF), estimated to occur in 1-5% of cases after myeloablative conditioning and in up to 30% of cases after reduced-intensity conditioning (RIC),1 still remains a relevant cause of morbidity and mortality after allogeneic hematopoietic stem cell transplantation (HSCT).2 Despite a slight reduction of its incidence over the last decade, mortality after GF remains as high as 11%.3 To date, in the absence of effective treatment options, re-transplantation, from either the same, or whenever possible, a different donor is considered the treatment of choice.2 Currently identified risk factors for GF include: i) human leukocyte antigen (HLA)-disparity and sex mismatch in the donor/recipient pair; ii) presence of donor-specific antibodies (DSA) in the recipient; iii) T-cell depletion (TCD) of the graft; iv) ABO-blood group mismatch; v) use of RIC; vi) a diagnosis of non-malignant disorders (in particular thalassemia, severe aplastic anemia, SAA, and hemophagocytic lymphohistiocytosis, HLH); vii) viral infections; viii) low nucleated cell dose in the graft; and ix) the use of myelotoxic drugs in the post-transplant period.1–4 In the last two decades, several groups have investigated immune-mediated GF. In particular, it has been shown that immune-mediated GF is mainly caused by host T and natural killer (NK) cells surviving the conditioning regimen, through a classical alloreactive immune response against non-shared, major (in case of HLA-partially-matched HSCT) or minor (in case of fully HLA-matched HSCT) histocompatibility antigens.2,5,6 However, to date the molecular pathways involved in immune-mediated GF have not yet been completely clarified. Indeed, since the inhibition of different pathways (including perforin-FasL−, TNFR-1−, and TRAIL-dependent cytotoxicity) did not prove to be efficient in preventing GF, the pathophysiological mechanisms responsible for GF seem to be multiple and likely to be redundant.7 Nonetheless, consistently over the years, different groups have suggested a pivotal pathogenic role of IFNγ in GF pathophysiology,8–14 through both direct [e.g. inhibition of hematopoietic stem cell (HSC) self-renewal, proliferative capacity, and multilineage differentiation]10,11 and indirect (e.g. induction of FAS expression on HSC, with increased apoptosis in the presence of activated cytotoxic T cells)8,12 effects. Despite these experimental data, there has still not been any in vivo characterization of GF in humans. Indeed, although the expansion of host CD8+ T cells in patients experiencing GF has been previously demonstrated in vivo,15,16 a more detailed characterization of this cell population is lacking. Thus, we started a prospective study aimed at better characterizing the pathophysiology of GF, focusing on the identification of biological markers that: (i) could predict early the occurrence of GF in the clinical setting; and (ii) could be used as a therapeutic target with clinically available biological agents. For this purpose, we broadly investigated cytokine and chemokine levels in peripheral blood (PB), as well as the cellular features in bone marrow (BM) biopsies of patients experiencing this complication. After confirming in vivo a role of IFNγ-pathway in the development of GF, we also investigated in an animal model of GF whether the sole inhibition of IFNγ would be able to prevent/treat GF. Finally, in view of these findings and the similarity between immune-mediated GF and HLH, we treated, in compassionate use (CU), with emapalumab, an anti-IFNγ monoclonal antibody recently approved for the treatment of HLH,17 three patients with primary HLH, who, after having experienced GF, underwent a second HSCT. Methods Patients Patients aged from 0.3 to 21 years, who received an allograft from any type of donor/stem cell source between January 1st 2016 and August 31st 2017 at the IRCCS Bambino Gesù Children’s Hospital in Rome, Italy, were considered eligible for the study. All patients or legal guardians provided written informed consent, and the entire research was conducted under institutional review board approved protocols and in accordance with the Declaration of Helsinki. The Bambino Gesù Children’s Hospital Institutional Review Board approved the study. Cytokine profile In order to identify a cytokine/chemokine profile predictive of GF, PB samples were collected at different time points after HSCT: day 0, +3±2, +7±2, +10±2, +14±2, +30±2 after transplantation. Validated MesoScale Discovery (MSD, Rockville, MD, USA) platform-based immunoassay was used for the quantification of IFNγ, sIL2Rα, CXCL9, CXCL10, TNFα, IL6, IL10, and sCD163 serum levels. Bone marrow biopsy: histopathology analysis and immunofluorescence Bone marrow biopsies were obtained when GF was suspected. (Since BM characterization was a secondary end point of this study and BM aspiration is not routinely performed in this condition, parents/legal guardians could refuse the procedure.) Details on BM specimen preparation, histopathology analysis and immunofluorescence are reported in the Online Supplementary Appendix. Immune-phenotypic analysis The following monoclonal antibodies (mAbs) were used: anti-CD3, CD4, CD8, CD25, CD27, CD28, CD45RA, CD45RO, CD56, CD57, CD62L, CD95, CD127, CD137, CD197, CD223 (Lag3), CD279 (PD1), and CD366 (TIM3) (BD Biosciences, NJ, Biolegend, CA and Affymetrix, CA, USA). In vivo murine model of hematopoietic stem cell transplantation rejection C57BL/6 Ifngr1−/− mice were used as recipient, while C57BL/6 Ifngr1+/+ were used as donor. All animal experiments were performed in accordance with the Swiss animal protection law. Details on experiments are reported in the Online Supplementary Appendix. Emapalumab administration in compassionate use to hemophagocytic lymphohistiocytosis patients experiencing graft failure Emapalumab (previously known as NI-0501), a fully human anti-IFNγ monoclonal antibody, was administered on a CU basis (after local ethical committee approval) to three patients affected by HLH who experienced GF after a first TCD HSCT from a partially-matched family donor (PMFD) with the aim of preventing flares of HLH and a second GF. The drug was administered by 1-hour intravenous infusion twice a week until sustained donor engraftment or GF. The dose varied between 1 and 6 mg/kg, based on pharmacokinetic data. Additional methods are presented in the Online Supplementary Appendix. Statistical analysis Unless otherwise specified, quantitative variables were reported as Mean±Standard Error of Mean (SEM); categorical variables were expressed as absolute value and percentage. Clinical characteristics of patients were compared using the χ2 test or Fisher exact test for categorical variables, while the Mann-Whitney rank sum test or the Student t-test (two-sided) was used for continuous variables, as appropriate. For multiple comparison analyses, statistical significance was evaluated by a repeated measure ANOVA test, followed by a Log-rank (Mantel-Cox) test for multiple comparisons. Results Patients’ characteristics During the study period, 15 consecutive patients who experienced GF were eligible for the study. Most of them were affected by non-malignant disorders characterized by a high risk of GF (e.g. SAA and HLH) and received a TCD allograft from a PMFD. Fifteen children, matched for transplant characteristics, who had sustained donor engraftment during the same period were used as controls. Patients’ and control characteristics are detailed in Table 1. Main transplant characteristics (i.e. conditioning regimen, type of donor, graft manipulation) were comparable between the two groups (except for a trend for a lower age in the GF group). Of the 15 patients experiencing GF, ten were tested for anti-HLA antibodies, which were detected in five patients (50%). Those who had a mean fluorescence intensity (MFI) of anti-HLA antibodies >5000 received rituximab and underwent plasma-exchange to lower the value below the threshold of 5000 MFI;18 this treatment successfully reduced the MFI value in all cases. Table 1. Characteristics of patients who either did or did not experience graft failure (GF). Signs and symptoms of patients who either did or did not experience GF are detailed in Table 2. The most frequent sign associated with GF was fever, occurring at a median time of six days from the infusion of the graft (range 1-16 days). Moreover, both lactate dehydrogenase (LDH) and ferritin increased in many patients (80% and 46.7%, respectively); these laboratory findings appeared late after HSCT (at a median of 11 and 10 days, respectively). All patients received steroids in an attempt to avoid GF, without benefit. Chimerism analysis performed on PB showed only recipient cells in all GF cases, while in all controls but one, who showed mixed chimerism, only donor-origin cells were found. Table 2. Signs and symptoms of patients who experienced graft failure (GF). Cytokine/chemokine profile Kinetics of IFNγ, CXCL9, IL10 and IL2Rα serum levels are shown in Figure 1A-D, while serum levels of TNFα, CXCL10, sCD163 and IL6 are shown in Figure 2A-D. Serum levels of these cytokines/chemokines differed between patients experiencing GF and controls, starting from the first days after the infusion of the graft. Notably, for IFNγ, CXCL9, IL10 and TNFα, this difference became statistically significant already at day +3 after HSCT. In particular, mean IFNγ levels at day +3 were 8859±7502 pg/mL in GF patients versus 0 pg/mL in controls (P=0.03); CXCL9 levels were 1514.0±773 pg/ml versus 233.6±50.1 pg/mL (P=0.0006); IL10 levels were 58.8±39.1 pg/mL versus 1.7±1.1 pg/mL (P=0.01); TNFα levels were 3.5±1.0 pg/mL versus 0.9±0.2 pg/mL (P=0.02). In this cohort, receiver operating characteristics (ROC) analysis on CXCL9 levels at day +3 showed an area under the curve (AUC) of 0.905 [95% Confidence Interval (CI) 0.709-0.987; P<0.0001] (Online Supplementary Figure S1); a cut-off value of 274.5 pg/mL had a sensitivity of 88.89% and a specificity of 78.57%. The ROC analysis of other markers, which were significantly increased at day +3 showed an AUC of 0.802 for TNFα (95%CI: 0.566-0.944; P=0.006), of 0.756 for IL10 (95%CI: 0.529-0.912; P=0.011) and of 0.682 for IFNγ (95%CI: 0.471-0.849; P=0.017). Figure 1. Cytokine/chemokine profile. Serum levels of interferon (IFN)-γ (A), CXCL9 (B), CXCL10 (C), and sIL2Rα (D) in patients who either did (red line) or did not (blue line) experience graft failure (GF). All graphs represent Mean and Standard Error of Mean for each variable. HSCT: hematopoietic stem cell transplantation. Figure 2. Cytokine/chemokine profile. Serum levels of TNFα (A), CXCL10 (B), sCD163 (C), IL6 (panel D). Red line: patients who experience graft failure (GF); blue line: controls. All graphs represent Mean and Standard Error of Mean for each variable. HSCT: hematopoietic stem cell transplantation. Since primary HLH patients commonly present increased IFNγ and its related chemokines serum levels during disease reactivation/flare (that is frequent after failure of HSCT19), we performed additional analyses excluding this subset of patients in order to validate the data in disorders other than HLH. Even after excluding HLH patients, CXCL9 and IL10 serum levels remained significantly higher in patients experiencing GF in comparison with controls (Online Supplementary Figure S2). Activation of macrophages and T lymphocytes characterizes graft failure in allogeneic hematopoietic stem cell transplantation Bone marrow biopsies were obtained at time of GF in seven patients and were compared to those of five controls (obtained in a similar time period, i.e. between 2 and 3 weeks after HSCT). In all GF patients, evaluation of BM morphology showed different stages of GF with reduced cellularity (Figure 3A and Online Supplementary Figure S3A and B) as compared to patients with sustained donor engraftment (Online Supplementary Figure S4A). In GF patients, the percentage of myelocytes and erythroid precursors was reduced compared to controls (Figure 3B). Erythroid colonies were markedly smaller, with a higher percentage of premature erythroid cells. The megakaryocytic lineage was well represented in all GF cases, but with irregular distribution (Figure 3C). In several areas of the specimens, a remarkable number of apoptotic cells partially grouped in clusters was observed (Figure 3D). All biopsies showed stromal damage resulting in edema (Figure 3E). While the total number of CD68+ macrophages was comparable between GF patients and controls (Figure 4A), significantly higher percentages of CD68+ and CD163+ macrophages, with cellular fragments, erythrocytes and lipid vacuoles in their cytoplasm, (indicating activation and phagocytic activity) (Figure 3F and G and Online Supplementary Figure S3C and D), were observed in comparison to controls [median 80% (range 30-100%) vs. 0% (range 0-5%); P<0.0001] (Figure 4B and Online Supplementary Figure S4B and C). In all analyzed samples from GF patients, a significant increase in T lymphocytes (Figures 3H and 4A and Online Supplementary Figure S3G), with a predominance of CD8+ cytotoxic T cells, expressing perforin, Granzyme B and TIA-1 (Figures 3I and J and 4A and Online Supplementary Figure S5) was observed. The Online Supplementary Appendix provides further details. Figure 3. Immunohistochemistry evaluation of bone marrow (BM) specimens in a patient experiencing graft failure (Pt #4). (A) Hematoxylin & eosin (H&E) staining of a BM specimen at 4X magnification. (B) Evaluation of erythroid colony spreading by glycophorin staining (10X). (C) Megakaryocyte distribution evaluated by CD61 expression (10X). (D) H&E staining at 40X showing apoptotic events. (E) H&E staining revealing stromal damage and edema development (40X). Characterization of the macrophage population by CD68 (F) and CD163 (G) staining (40X). Characterization and distribution of T lymphocytes by analysis of CD3 (H), CD4 (I), and CD8 (J) expression (10X). Figure 4. Immunohistochemistry characterization of bone marrow (BM) in patients who either did or did not experience graft failure (GF). (A) Comparison of absolute number of CD3+, CD4+, CD8+, CD68+, TIA-1+, perforin+ and granzyme+ cells in BM of GF patients and controls (CTRL). The total number of positive cell for each marker was counted in five fields per sample under 20-fold magnification and reported as Mean±Standard Deviation. (B) Percentages of CD68+ cells with hemophagocytic activity (i.e. showing cellular fragments, erythrocytes and lipid vacuoles in their cytoplasm) in BM of GF patients and CTRL. *P<0.05; **P<0.01; ***P<0.001. Polyclonal T-cell pattern with predominant CD8 effector memory phenotype effector memory phenotype In order to better characterize the role of T lymphocytes in GF, the TCR repertoire was initially analyzed in the CD3+ population, showing a polyclonal distribution of the Vβ chains (Online Supplementary Figure S6). Then, we extended our analysis on BM-infiltrating lymphocytes through flowcytometry in both controls and GF patients. Regarding NK (CD56+/CD3−) and γδ T cells (CD3+/CD4−/CD8−) no difference was observed between the two patient groups (data not shown). By contrast, in the αβ T-cell subset, the analysis revealed a significant difference in both CD4 (58.9%±13.4% vs. 7.6%±7.3%, controls vs. GF patients) and CD8 (25.9%±6.1% vs. 66.5%±18.2%, controls vs.GF patients) subsets (P<0.0001 and P=0.0018, respectively) (Figure 5A). We further characterized both CD4+ and CD8+ populations for the expression of memory markers. While no significant difference was detected in the CD4+ subpopulation, the CD8+ subset displayed a significant enrichment of effector memory T cells (EfM) (CD45RO+/CCR7-) (40.3±24.6% vs. 20.7%±7.3%, GF patients vs. CTRL patients; P=0.034) (Figure 5B and C) and a significant reduction of the naïve subset (CD45RA+/CCR7+) (18.6%±16.6% vs. 28.6%±12.1%, GF patients vs. controls; P=0.014). See Online Supplementary Appendix for further details. Figure 5. Immuno-characterization of the T lymphocytes present in bone marrow aspirates of patients who either did or did not experience graft failure (GF). (A) Flow cytometry analysis of CD4+ and CD8+ population in patients with GF and controls (CTRL). Distribution of naïve (CD45RA+/CCR7+), central memory (CD45RO+/CCR7+), effector memory (CD45RO+/CCR7−), effector terminal (CD45RA+/CCR7−), and NK-T (CD3+/CD56+) subsets in CD4+ (B) or CD8+ (C) T cells. Activation and exhaustion profile in both the CD4+ and CD8+ population by the analysis of CD95 (D), CD127 (E), and CD57 (F). (A, D, E, and F) Each patient or CTRL is represented by a symbol and a horizontal line marks the median. (B and C) The average (+) and Median±Standard Deviation are shown. *P<0.05; **P<0.01; ***P<0.001; ****P<0.0001. Increasing expression of activation and exhaustion markers on T cells during graft failure We evaluated the expression of several activation and exhaustion markers on infiltrating cells. As expected, in patients experiencing GF, both CD4+ and CD8+ cells displayed a significant activation profile, as demonstrated by the overexpression of CD95 (69.2%±23.0% vs. 93.9%±6.9% and 57.9%±27.2% vs. 98.35%±2.0%, controls vs. GF patients, respectively; P=0.021 and P=0.002) (Figure 5D) and downregulation of CD127 (recently shown to be associated with prolonged T-cell receptor stimulation20) on the proliferating CD8+ cells (69.3%±16.9% vs. 37.9%±18.8%, controls vs. GF patients, respectively; P=0.014) (Figure 5E). The expression of several exhaustion and senescence markers confirmed the status of prolonged activation of T lymphocytes located in the BM of GF patients, such as the upregulation of CD57 (CD57+: 10.2%±10.5% vs. 37.4%±12.4% and 34.7%±17.3% vs. 68.0%±18.8% controls vs. GF patients in CD4 and CD8 respectively; P=0.003 and P=0.011) (Figure 5F). See Online Supplementary Appendix for further details. Interferon-γ drives rejection of donor cells in Ifngr1−/–mice In order to understand if the sole IFNγ-inhibition would be sufficient to prevent GF, we used an established mouse model of GF.13 As previously reported by Rottman et al.,13 the infection of Ifngr1−/− mice with Bacillus Calmette– Guérin (BCG) resulted in a rapid increase of circulating IFNγ levels reaching a concentration of 11,000 pg/mL on day 20 post-infection (Figure 6A). HSCT performed at day 21, i.e. at the peak of IFNγ levels, resulted in poor chimerism as only 5% of the Ifngr1+/+ donor cells engrafted in the BCG-infected Ifngr1−/− recipient mice. After day 21 post-BCG infection, serum IFNγ levels gradually decreased to a steady state level of approximately 100 pg/mL. This decrease in IFNγ serum levels correlated with an increase in chimerism as the Ifngr1−/− recipient mice exhibited 19% HSC engraftment of donor cells at day 84 (Figure 6A). For further assessing the role played by IFNγ in GF, BCG-infected Ifngr1−/− recipient mice were given a neutralizing IFNγ mAb, XMG1.2, pre-and post-HSCT. Neutralization of IFNγ improved engraftment in BCG-infected Ifngr1−/−recipient mice because, at three months after the allograft, 45% of the lymphocytes were of donor origin (i.e. Ly5.1 positive), as compared to 19% in isotype control-treated mice (Figure 6B). In order to assess IFNγ activity and ensure neutralization by XMG1.2, the IFNγ-dependent chemokine CXCL9 was measured. A decrease in CXCL9 serum levels during the XMG1.2 treatment was observed, confirming IFNγ neutralization in contrast to isotype control-treated mice (Figure 6C). Once XMG1.2 treatment was interrupted, at day 42 post-BCG infection, a gradual increase in CXCL9 serum levels was observed, indicating restoration of IFNγ activity. Figure 6. Successful hematopoietic stem cell transplantation (HSCT) chimerism in interferon (IFN)-γR1−/− mice correlates with low IFNγ activity; circulating CXCL9 levels is a biomarker of in vivo IFNγ activity. Ifngr1−/− mice (expressing the Ly5.2 congenic marker) were intravenously (i.v.) infected with 1,106 CFU of Bacillus Calmette–Guérin (BCG) (strain Pasteur 1173P2). After 14, 20, 28, 35 and 42 days mice were treated i.v. with 100 mg/kg of an isotype control (n=5) or the anti-mIFNγ, XMG1.2 (n=5). At day 21, mice were infused with bone marrow from Ifngr+/+ mice, expressing the Ly5.1 marker, after mild irradiation (550 rads). Chimerism, assessed by determining the surface expression of Ly5.1 and Ly5.2 on lymphocytes, was analyzed by flow cytometry at different time points after HSCT treatment. IFNγ levels were quantified at different time points by ELISA using the Luminex technology. (A) Graph represents the super-imposition of the chimerism (black straight line) and the IFNγ levels (gray dotted line) in the isotype control treated mice. (B) Graph represents the chimerism determined in mice treated with the isotype control (black straight line) or with the XMG1.2 (gray straight line) mAbs. (C) Ifngr1−/− mice were i.v. infected with 1.106 CFU of BCG (strain Pasteur 1173P2). After 14, 20, 28, 35 and 42 days mice were treated i.v. with 100 mg/kg of an isotype control (black straight line; n=5) or the anti-mIFNγ, XMG1.2 (gray straight line; n=5). At day 21, mice were transplanted with bone marrow from Ifngr+/+ mice, expressing the Ly5.1 marker, after mild irradiation (550 rads). At different time points post-BCG infection, circulating CXCL9 levels were quantified by ELISA using the Luminex technology. Ab: antibody. Emapalumab administration to patients after hematopoietic stem cell transplantation failure Three patients with primary HLH who experienced GF together with disease reactivation after a first TCD HSCT from a PMFD were treated with emapalumab both before and after the second HSCT (details are reported in Online Supplementary Table S1). For all these patients, the use of the other parent as a donor was not possible because of non-eligibility due to viral hepatitis. The CU of emapalumab was requested and obtained with the objective of controlling, without the use of myelosuppressive drugs other than those used in the conditioning regimen, HLH reactivation before and after a second HSCT. Emapalumab was administered at doses of 1-6 mg/kg every three days. Drug infusions were well tolerated and no significant safety event occurred. Two patients engrafted, while one rejected also the second HSCT without, however, experiencing a new HLH flare. This patient was successfully rescued with a third HSCT employing an unrelated cord blood (UCB) unit (notably, she received emapalumab until 3 days before UCB infusion). Remarkably, the two patients who engrafted upon treatment with emapalumab had very low levels of CXCL9 (i.e. below 102 pg/mL), indicating IFNγ neutralization, while this was not the case for the third patient at the time of the second transplant rejection. All these three patients are currently alive and disease-free, with a follow up of 24, 23 and 21 months, respectively. Discussion Diagnosis and treatment of GF in HSCT recipients remain challenging. Indeed, sign and symptoms (e.g. fever, increase in LDH or ferritin serum levels) associated with this transplant complication are non-specific; moreover, re-transplantation, although associated with relevant risk of tissue-toxicity and infections, represents the treatment of choice, since steroids and other immunosuppressive drugs are usually ineffective for rescuing these patients.2 In this study, we investigated humoral and cellular features of GF occurring after allogeneic HSCT in children, documenting a pivotal role played by IFNγ in the pathophysiology of this complication. Apart from the indirect evidence provided by the observation of very high rates of primary and secondary rejection after HLA-identical HSCT in patients with IFNγ-receptor 1 deficiency,21 currently available clinical data about the role of IFNγ in GF in humans remain limited. Interestingly, we found that GF is characterized by the same clinical (including high-grade fever, hepato/splenomegaly, hemophagocytosis in BM)22,23 and laboratory (i.e. increased ferritin, IFNγ, CXCL9, CXCL10, sCD163 and sIL-2Rα levels)24–28 features found in patients with HLH, where a central role of IFNγ has been shown.29 Our data indicate that IFNγ levels, and even more CXCL9 levels measured in PB, can predict GF with high sensitivity and specificity already at day +3 after graft infusion, while signs and symptoms of GF appear only later (see Table 2). Indeed, the current proposed risk score for GF determined on day +21 after HSCT, based on eight patient and transplant variables, showed good specificity, but low sensitivity.1 The high accuracy of CXCL9 in predicting GF, as indicated by the AUC of 0.905, renders this chemokine an ideal “candidate biomarker”, as stated by the 2014 National Institutes of Health consensus on biomarkers.30,31 CXCL9, also known as monokine induced by γ-interferon (MIG), is a chemokine specifically induced by IFNγ,32 and represent the most sensitive and specific of the soluble factors we analyzed. It binds to the chemokine receptor CXCL3 expressed on naïve T cells, Th1 CD4+ T cells, effector CD8+ T cells, as well as on NK and NKT cells, driving Th1 inflammation. Circulating CXCL9 levels have been shown to reflect the amount of IFNγ produced in organs, such as liver and spleen,25 which are the typical target of inflammation. This strong correlation with IFNγ produced in organs rather than in blood provides an explanation why, despite high CXCL9 serum levels, serum levels of IFNγ were found to be low or even undetectable in a few of our GF patients. Furthermore, elevated levels of CXCL9 have been related to graft rejection in solid organ transplantation (such as heart, kidney and lung transplantation),33–35 but, to the best of our knowledge, this is the first report demonstrating that the hyperproduction of IFNγ in GF occurring after HSCT results in increased CXCL9 serum levels. Among other cytokines/chemokines, we also observed increased levels of IL10, an important Th2 cytokine with anti-inflammatory properties, this finding being in agreement with the hyperproduction of this molecule recorded in patients with HLH.36 Our results are not only relevant for diagnostic purposes, but also suggest that IFNγ is a potential therapeutic target in GF. Indeed, independently of the mechanism of IFNγ-mediated GF (i.e. direct effect on HSC or HLH-like effect), our results support the investigation of IFNγ neutralization for prevention and/or treatment of GF in patients undergoing HSCT. The encouraging efficacy and safety data reported from the ongoing study in primary HLH with emapalumab (NI-0501), an anti-IFNγ monoclonal antibody,17,37 provides additional support for the rationale for using this drug.38 The data we generated in the murine model of GF confirm and extend the role played by IFNγ previously demonstrated by Rottman et al.13 Moreover, we also show that the sole neutralization of IFNγ, without the administration of anti-IL12 (employed in the experiments reported by Rottman et al.),13 is able to improve engraftment. The observation that decreased CXCL9 production correlates with improved HSCT chimerism provides further support to a therapeutic intervention aimed at neutralizing IFNγ-pathway signaling. Finally, the data obtained in the three patients treated on a CU basis indicate that the use of an anti-IFNγ monoclonal antibody is safe also in a very fragile population, namely infants with a previous GF undergoing a second HSCT. Four out of the seven patients we studied who underwent BM aspirate and biopsy showed evidence of hemophagocytosis. Indeed, it has been shown that an increased number of hemophagocytic macrophages in the BM obtained 14±7 days after HSCT is associated with higher risk of death due to GF.39 Moreover, in a cohort of adult patients receiving cord blood transplantation, GF was strictly related to the occurrence of HLH manifestations.23 Recently, in a retrospective study on peri-engraftment BM samples from 32 adult patients, Kawashima et al. proposed two histological measures, namely macrophage ratio and CD8+ ratio (defined as the ratio between the macrophage or CD8+ lymphocyte number on the total nucleated cell number), as predictors of GF at day +14.15 Despite some preliminary studies characterizing host T cell expansion in patients with GF,15,16 no information is available regarding the phenotype of these cells. Our data indicate an active role of T lymphocytes in mediating GF. As previously reported,15 in these patients, the mononuclear infiltrate is mainly constituted by cytotoxic CD8+ lymphocytes with a predominant effector memory phenotype. This population was demonstrated to be activated, proliferating and cytotoxic, expressing specific molecules, such as Granzyme B, Perforin and TIA-1, involved in target-killing, as well as various activation and proliferation markers. Interestingly, we observed that CD8+ lymphocyte expansion is predominantly polyclonal, suggesting that the immune response is directed towards several antigens and not against few immunodominant epitopes. However, a significant enrichment of certain β clones was found. The cytopathic effect was clearly demonstrated by apoptotic cells surrounding proliferating T cells, which are long-term activated, as demonstrated by the expression of several exhaustion markers.40,41 Furthermore, the remaining γ/d and CD4+ T-cell populations are similarly expressing exhaustion markers, underlying an over-stimulated environment. Notably, a particular behavior was observed in the NKT-cell population with a significant reduction of CD8+ NKT, probably due to their activation and a significant increase of CD4+ NKT. The role of these cells is yet to be fully elucidated, although they were shown to be able to prevent pancreatic islet transplant rejection, but also to sustain CD8+ T-cell expansion.42,43 Given these data, a treatment able to interrupt the overproduction of molecules responsible for inflammation,33 such as an anti-IFNγ, could be beneficial in this setting. Fifty percent of tested patients had anti-HLA antibodies: all those with positivity >5,000 MFI received a desensitization therapy in order to lower the antibody title with the aim of reducing the risk of GF. Although we cannot exclude a role of anti-HLA antibodies in causing GF in our patients, all five positive patients showed increased values of IFNγ and/or related cytokines after HSCT. Thus, we can hypothesize that there may be a common final pathway and/or combined action (like that reported in solid organ transplantation)44 between humoral and cellular mechanisms sustaining GF. Limitations of this study are the lack of a validation cohort and the relatively small number of patients included in the study. Another important limitation is that most patients experiencing GF that we report were transplanted from a PMFD after a TCD procedure (both being well-known risk factors for GF);2,3 thus, our results should be further validated in other transplant settings, especially when post-transplant pharmacological graft-versus-host disease prophylaxis is used. Indeed, the use of calcineurin inhibitors or other immunosuppressive agents can modify IFNγ (and related cytokines) secretion kinetics.45 Overall, our data suggest that immune-mediated GF may share clinical and laboratory characteristics with HLH. Besides providing evidence for further investigating the use of markers to allow a non-invasive, prompt identification of patients at high risk of developing this severe complication of HSCT, the increased serum levels of IFNγ and CXCL9 found in GF patients provide a rationale for investigating a targeted therapy (i.e. anti-IFNγ therapy) in this complication. We are currently designing a clinical trial on the use of emapalumab for prevention and/or treatment of GF in patients at high risk of developing this complication. Footnotes Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/104/11/2314 Funding This work was supported by “Ricerca corrente” (Ministero della Salute) (PM), Investigator Grant 2015 Id. 17200 by Associazione Italiana per la Ricerca sul Cancro (AIRC) (FL) and by Novimmune SA, Switzerland. Received January 7, 2019. Accepted February 18, 2019. Copyright© 2019 Ferrata Storti Foundation Material published in Haematologica is covered by copyright. All rights are reserved to the Ferrata Storti Foundation. Use of published material is allowed under the following terms and conditions: https://creativecommons.org/licenses/by-nc/4.0/legalcode. Copies of published material are allowed for personal or internal use. Sharing published material for non-commercial purposes is subject to the following conditions: https://creativecommons.org/licenses/by-nc/4.0/legalcode, sect. 3. Reproducing and sharing published material for commercial purposes is not allowed without permission in writing from the publisher. References 1.↵Olsson RF, Logan BR, Chaudhury S, et al. Primary graft failure after myeloablative allogeneic hematopoietic cell transplantation for hematologic malignancies. Leukemia. 2015;29(8):1754–1762.OpenUrlCrossRefPubMed 2.↵Locatelli F, Lucarelli B, Merli P. Current and future approaches to treat graft failure after allogeneic hematopoietic stem cell transplantation. Expert Opin Pharmacother. 2014;15(1):23–36.OpenUrlCrossRefPubMed 3.↵Olsson R, Remberger M, Schaffer M, et al. Graft failure in the modern era of allogeneic hematopoietic SCT. Bone Marrow Transplant. 2013;48(4):537–543.OpenUrlCrossRefPubMed 4.↵Cluzeau T, Lambert J, Raus N, et al. Risk factors and outcome of graft failure after HLA matched and mismatched unrelated donor hematopoietic stem cell transplantation: a study on behalf of SFGM-TC and SFHI. Bone Marrow Transplant. 2016;51(5):687–691.OpenUrl 5.↵Masouridi-Levrat S, Simonetta F, Chalandon Y. Immunological Basis of Bone Marrow Failure after Allogeneic Hematopoietic Stem Cell Transplantation. Front Immunol. 2016;7:362.OpenUrlCrossRef 6.↵Murphy WJ, Kumar V, Bennett M. Acute rejection of murine bone marrow allografts by natural killer cells and T cells. Differences in kinetics and target antigens recognized. J Exp Med. 1987;166(5):1499–1509. 7.↵Komatsu M, Mammolenti M, Jones M, Jurecic R, Sayers TJ, Levy RB. Antigen-primed CD8+ T cells can mediate resistance, preventing allogeneic marrow engraftment in the simultaneous absence of perforin-CD95L-TNFR1-and TRAIL-dependent killing. Blood. 2003;101(10):3991–3999. 8.↵Chen J, Feng X, Desierto MJ, Keyvanfar K, Young NS. IFN-γ-mediated hematopoietic cell destruction in murine models of immune-mediated bone marrow failure. Blood. 2015;126(24):2621–2631. 9.Chen J, Lipovsky K, Ellison FM, Calado RT, Young NS. Bystander destruction of hematopoietic progenitor and stem cells in a mouse model of infusion-induced bone marrow failure. Blood. 2004;104(6):1671–1678. 10.↵de Bruin AM, Demirel O, Hooibrink B, Brandts CH, Nolte MA. Interferon-γ impairs proliferation of hematopoietic stem cells in mice. Blood. 2013;121(18):3578–3585. 11.↵Lin FC, Karwan M, Saleh B, et al. IFN-γ causes aplastic anemia by altering hematopoietic stem/progenitor cell composition and disrupting lineage differentiation. Blood. 2014;124(25):3699–3708. 12.↵Maciejewski J, Selleri C, Anderson S, Young NS. Fas antigen expression on CD34+ human marrow cells is induced by interferon γ and tumor necrosis factor α and potentiates cytokine-mediated hematopoietic suppression in vitro. Blood. 1995;85(11): 3183–3190. 13.↵Rottman M, Soudais C, Vogt G, et al. IFN-γ mediates the rejection of haematopoietic stem cells in IFN-γR1-deficient hosts. PLoS Med. 2008;5(1):e26.OpenUrlCrossRefPubMed 14.↵Selleri C, Maciejewski JP, Sato T, Young NS. Interferon-γ constitutively expressed in the stromal microenvironment of human marrow cultures mediates potent hematopoietic inhibition. Blood. 1996; 87(10):4149–4157. 15.↵Kawashima N, Terakura S, Nishiwaki S, et al. Increase of bone marrow macrophages and CD8+ T lymphocytes predict graft failure after allogeneic bone marrow or cord blood transplantation. Bone Marrow Transplant. 2017;52(8):1164–1170.OpenUrl 16.↵Koyama M, Hashimoto D, Nagafuji K, et al. Expansion of donor-reactive host T cells in primary graft failure after allogeneic hematopoietic SCT following reduced-intensity conditioning. Bone Marrow Transplant. 2014;49(1):110–115.OpenUrl 17.↵Jordan M, Locatelli F, Allen C, et al. A Novel Targeted Approach to the Treatment of Hemophagocytic Lymphohistiocytosis (HLH) with an Anti-Interferon γ (IFN γ) Monoclonal Antibody (mAb), NI-0501: First Results from a Pilot Phase 2 Study in Children with Primary HLH. Blood. 2015; 126(23):3.OpenUrl 18.↵Ciurea SO, Thall PF, Milton DR, et al. Complement-Binding Donor-Specific Anti-HLA Antibodies and Risk of Primary Graft Failure in Hematopoietic Stem Cell Transplantation. Biol Blood Marrow Transplant. 2015;21(8):1392–1398.OpenUrlCrossRefPubMed 19.↵Messina C, Zecca M, Fagioli F, et al. Outcomes of Children with Hemophagocytic Lymphohistiocytosis Given Allogeneic Hematopoietic Stem Cell Transplantation in Italy. Biol Blood Marrow Transplant. 2018;24(6):1223–1231.OpenUrl 20.↵Utzschneider DT, Alfei F, Roelli P, et al. High antigen levels induce an exhausted pheno type in a chronic infection without impairing T cell expansion and survival. J Exp Med. 2016;213(9):1819–1834. 21.↵Roesler J, Horwitz ME, Picard C, et al. Hematopoietic stem cell transplantation for complete IFN-γ receptor 1 deficiency: a multi-institutional survey. J Pediatr. 2004; 145(6):806–812. 22.↵Abe Y, Choi I, Hara K, et al. Hemophagocytic syndrome: a rare complication of allogeneic nonmyeloablative hematopoietic stem cell transplantation. Bone Marrow Transplant. 2002;29(9):799–801. 23.↵Takagi S, Masuoka K, Uchida N, et al. High incidence of haemophagocytic syndrome following umbilical cord blood transplantation for adults. Br J Haematol. 2009; 147(4):543–553.OpenUrlCrossRefPubMed 24.↵Bracaglia C, de Graaf K, Pires Marafon D, et al. Elevated circulating levels of interferon-γ and interferon-γ-induced chemokines characterise patients with macrophage activation syndrome complicating systemic juvenile idiopathic arthritis. Ann Rheum Dis. 2017;76(1):166–172. 25.↵Buatois V, Chatel L, Cons L, et al. Use of a mouse model to identify a blood biomarker for IFNγ activity in pediatric secondary hemophagocytic lymphohistiocytosis. Transl Res. 2017;180:37–52.e2.OpenUrl 26.Henter JI, Elinder G, Soder O, Hansson M, Andersson B, Andersson U. Hypercytokinemia in familial hemophagocytic lymphohistiocytosis. Blood. 1991; 78(11):2918–2922. 27.Xu XJ, Tang YM, Song H, et al. Diagnostic accuracy of a specific cytokine pattern in hemophagocytic lymphohistiocytosis in children. J Pediatr. 2012;160(6):984–990.e1. 28.↵Yang SL, Xu XJ, Tang YM, et al. Associations between inflammatory cytokines and organ damage in pediatric patients with hemophagocytic lymphohistiocytosis. Cytokine. 2016;85:14–17.OpenUrl 29.↵Jordan MB, Hildeman D, Kappler J, Marrack P. An animal model of hemophagocytic lymphohistiocytosis (HLH): CD8+ T cells and interferon γ are essential for the disorder. Blood. 2004;104(3):735–743. 30.↵Paczesny S. Biomarkers for posttransplantation outcomes. Blood. 2018;131(20):2193–2204. 31.↵Paczesny S, Hakim FT, Pidala J, et al. National Institutes of Health Consensus Development Project on Criteria for Clinical Trials in Chronic Graft-versus-Host Disease: III. The 2014 Biomarker Working Group Report. Biol Blood Marrow Transplant. 2015;21(5):780–792.OpenUrlCrossRefPubMed 32.↵Groom JR, Luster AD. CXCR3 ligands: redundant, collaborative and antagonistic functions. Immunol Cell Biol. 2011;89(2): 207–215. 33.↵Fahmy NM, Yamani MH, Starling RC, et al. Chemokine and chemokine receptor gene expression indicates acute rejection of human cardiac transplants. Transplantation. 2003;75(1):72–78. 34.Gupta A, Broin PO, Bao Y, et al. Clinical and molecular significance of microvascular inflammation in transplant kidney biopsies. Kidney Int. 2016;89(1):217–225.OpenUrlCrossRefPubMed 35.↵Medoff BD, Wain JC, Seung E, et al. CXCR3 and its ligands in a murine model of obliterative bronchiolitis: regulation and function. J Immunol. 2006;176(11):7087–7095. 36.↵An Q, Hu SY, Xuan CM, Jin MW, Ji Q, Wang Y. Interferon γ and interleukin 10 polymorphisms in Chinese children with hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2017;64(9). 37.↵Locatelli F, Jordan M, Allen C, et al. Safety and efficacy of emapalumab in pediatric patients with primary hemophagocytic lymphohistiocytosis. Blood. 2018;132(Suppl 1):LBA–6.OpenUrl 38.↵Prencipe G, Caiello I, Pascarella A, et al. Neutralization of interferon-γ reverts clinical and laboratory features in a mouse model of macrophage activation syndrome. J Allergy Clin Immunol. 2018;141(4):1439–1449.OpenUrl 39.↵Imahashi N, Inamoto Y, Ito M, et al. Clinical significance of hemophagocytosis in BM clot sections during the peri-engraftment period following allogeneic hematopoietic SCT. Bone Marrow Transplant. 2012; 47(3):387–394.OpenUrlPubMed 40.↵Ferris RL, Lu B, Kane LP. Too much of a good thing? Tim-3 and TCR signaling in T cell exhaustion. J Immunol. 2014;193(4):1525–1530. 41.↵Jin HT, Anderson AC, Tan WG, et al. Cooperation of Tim-3 and PD-1 in CD8 T-cell exhaustion during chronic viral infection. Proc Natl Acad Sci U S A. 2010; 107(33):14733–14738. 42.↵Ikehara Y, Yasunami Y, Kodama S, et al. CD4(+) Valpha14 natural killer T cells are essential for acceptance of rat islet xenografts in mice. J Clin Invest. 2000; 105(12):1761–1767. 43.↵Lin H, Nieda M, Rozenkov V, Nicol AJ. Analysis of the effect of different NKT cell subpopulations on the activation of CD4 and CD8 T cells, NK cells, and B cells. Exp Hematol. 2006;34(3):289–295.OpenUrlCrossRefPubMed 44.↵Zeglen S, Zakliczynski M, Wozniak-Grygiel E, et al. Mixed cellular and humoral acute rejection in elective biopsies from heart transplant recipients. Transplant Proc. 2009; 41(8):3202–3205.OpenUrlCrossRefPubMed 45.↵Grant CR, Holder BS, Liberal R, et al. Immunosuppressive drugs affect interferon (IFN)-γ and programmed cell death 1 (PD-1) kinetics in patients with newly diagnosed autoimmune hepatitis. Clin Exp Immunol. 2017;189(1):71–82.OpenUrl
|
|
Scooped by
Krishan Maggon
February 8, 2019 5:07 AM
|
The fact that the incidence is highest in people who are immunosuppressed provides some support for the idea that Merkel cell carcinoma is an immunogenic cancer, one that is related to immune function, and a good candidate for immunotherapy. The National Cancer Institute defines immunotherapy as “a type of therapy that uses substances to stimulate or suppress the immune system to help the body fight cancer, infection, and other diseases. Some types of immunotherapy only target certain cells of the immune system. Others affect the immune system in a general way. Types of immunotherapy include cytokines, vaccines, bacillus Calmette-Guerin (BCG), and some monoclonal antibodies.” Pembrolizumab is a monoclonal antibody. Other centers participating in the trial are University of Washington/Fred Hutchinson Cancer Research Center, Johns Hopkins Kimmel Cancer Center and Bloomberg–Kimmel Institute for Cancer Immunotherapy, Emory University, Moffitt Cancer Center, Mount Sinai Medical Center, University of California San Francisco, Yale University, Stanford University, University of Pittsburgh, Duke University Medical Center, Ohio State University Comprehensive Cancer Center, City of Hope, Fred Hutchinson Cancer Research Center/Cancer Immunotherapy Trials Network, University of Washington, and Axio Research. The research was supported by grants from the National Cancer Institute, the Merkel cell carcinoma (MCC) patient gift fund at University of Washington, the Kelsey Dickson MCC Challenge Grant from the Prostate Cancer Foundation, the Al Copeland Foundation, and Merck, which provided pembrolizumab and partial funding for this study.
|
|
Scooped by
Gilbert C FAURE
May 10, 2018 2:44 PM
|
Infections take their greatest toll in early life necessitating robust approaches to protect the very young. Here we review the rationale, current state and future research directions for one such approach: neonatal immunization. Challenges to neonatal immunization include natural concern about safety as well as a distinct neonatal immune system that is generally polarized against Th1 responses to many stimuli such that some vaccines that are effective in adults are not in newborns. Nevertheless, neonatal immunization could result in high population penetration as birth is a reliable point of healthcare contact, and offers an opportunity for early protection of the young, including preterm newborns who are deficient in maternal antibodies. Despite distinct immunity and reduced responses to some vaccines, several vaccines have proven safe and effective at birth. While some vaccines such as polysaccharide vaccines have little effectiveness at birth, hepatitis B vaccine (HBV) can prime at birth and requires multiple doses to achieve protection, whereas the live attenuated Bacille Calmette Guérin (BCG), may offer single shot protection, potentially in part via heterologous (“nonspecific”) beneficial effects. Additional vaccines have been studied at birth including those directed against pertussis, pneumococcus, Haemophilus influenza type B (Hib) and rotavirus providing important lessons. Current areas of research in neonatal vaccinology include characterization of early lif
|
|
Scooped by
Gilbert C FAURE
March 23, 2018 10:07 AM
|
Tuberculosis (TB) is a granulomatous disease that has affected humanity for thousands of years. The production of cytokines, such as IFN-γ and TNF-α, is fundamental in the formation and maintenance of granulomas and in the control of the disease. Recently, the introduction of TNF-α-blocking monoclonal antibodies, such as Infliximab, has brought improvements in the treatment of patients with chronic inflammatory diseases, but this treatment also increases the risk of reactivation of latent tuberculosis. Our objective was to analyze, in an in vitro model, the influence of Infliximab on the granulomatous reactions and on the production of antigen-specific cytokines (TNF-α, IFN-γ, IL-12p40, IL-10 and IL-17) from beads sensitized with soluble Bacillus Calmette-Guérin (BCG) antigens cultured in the presence of peripheral blood mononuclear cells (PBMC) from TB patients. We evaluated 76 individuals, with tuberculosis active, treated and subjects with positive PPD. Granuloma formation was induced in the presence or absence of Infliximab for up to 10 days. The use of Infliximab in cultures significantly blocked TNF-α production (p <0.05), and led to significant changes in granuloma structure, in vitro, only in the treated TB group. On the other hand, there was a significant reduction in the levels of IFN-γ, IL-12p40, IL-10 and IL-17 after TNF-α blockade in the three experimental groups (p <0.05). Taken together, our results demonstrate that TNF-α blockade by Infliximab directly influenced the structure of granuloma only in the treated TB group, but negatively modulated the production of Th1, Th17 and regulatory T cytokines in the three groups analyzed.
|
|
Scooped by
Gilbert C FAURE
July 29, 2015 7:47 AM
|
by Paulo Ranaivomanana, Vaomalala Raharimanga, Patrice M.
|









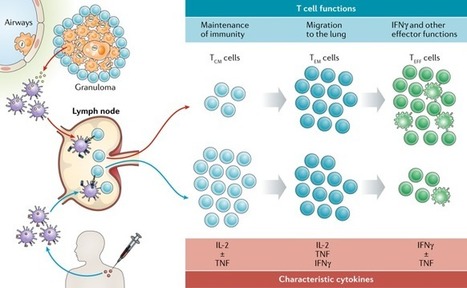
















https://www.scoop.it/topic/immunology-and-biotherapies?q=bcg