Your new post is loading...
Your new post is loading...

|
Scooped by
I2BC Paris-Saclay
July 26, 9:11 AM
|
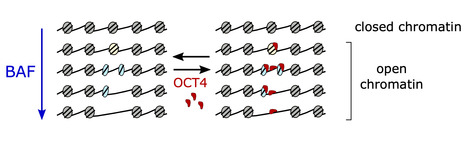
A tumor suppressor that generates subnucleosomes
Researchers from the I2BC, the CNRGH (CEA, National Center of Human Genomics Research), and the University of Edinburgh have revealed how the BAF chromatin remodeler changes the structure and accessibility of genetic material by producing subnucleosomal particles. These findings are published in Nature Structural and Molecular Biology. Mutations in genes encoding subunits of the BAF complex (BRG1/BRM-associated factors, also known as the SWI/SNF complex) are associated with 20% of human cancers, making this complex the second most frequently mutated factor after the p53 tumor suppressor.
The human genome exists in cells in the form of chromatin, composed of nucleosomes that condense and protect the genetic material. The nucleosomes represent a physical barrier for transcription factors and molecular machinery that regulate gene expression. The BAF complex is known for controlling chromatin opening at gene transcription regulatory elements. Until now, it was believed that the role of the BAF complex was to dissociate the nucleosomes to allow the transcription factors to access DNA.
Using a method that allows them to differentiate histone-free DNA fragments from nucleosomes and histone-containing particles smaller than nucleosomes (subnucleosomes), researchers coordinated by Matthieu Gérard (I2BC) revealed that the BAF complex facilitates the recruitment of the master transcription factor OCT4 using two distinct strategies: i) by generating the well-known histone-free DNA regions, which OCT4 binds if its target DNA motif is present; and ii) by producing a new class of subnucleosomal particles containing 50 to 80 base pairs associated with histones.
They show that the subnucleosomes act as a recruitment platform for OCT4 independently of the presence of its DNA motif. They identify in this publication a molecular mechanism in which the interaction between OCT4 and the subnucleosomes leads to a spectacular expansion of the OCT4 genomic binding interval. This mechanism aims to project OCT4 activity in chromatin opening within a genomic interval up to one order of magnitude larger than the interval bound by OCT4 on histone-free DNA.
This study suggests two main determinants for recruiting transcription factors onto the mammalian genome: the genetic determinant, the transcription factor motif within histone-free DNA, and an epigenetic determinant, which corresponds to the subnucleosomes produced by the BAF complex. More information: https://www.nature.com/articles/s41594-024-01344-0 Contact: Matthieu GERARD matthieu.gerard@i2bc.paris-saclay.fr
|
|
Scooped by
I2BC Paris-Saclay
July 2, 10:27 AM
|
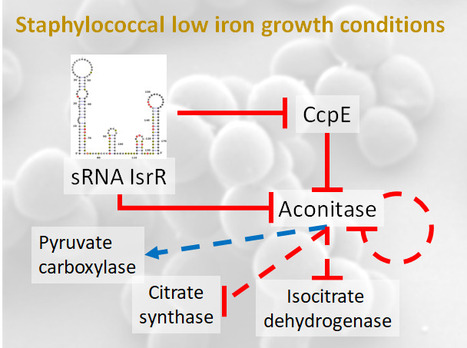
Staphylococcus aureus in the battle for iron: three post-transcriptional regulations at work
A feed-forward loop controlled by the regulatory sRNA IsrR and the aconitase moonlighting activity prevent aconitase production during iron deficiency. Staphylococcus aureus, responsible for nosocomial infections and septicemia, is the leading cause of bacterial mortality in most countries of the world. Its pathogenicity relies on its adaptation to a wide range of biotopes.
As iron bioavailability is sometimes a limiting factor for growth, bacteria have developed strategies to scavenge iron and reduce its utilization under iron-depleted growth conditions. One such strategy involves the modulation of iron-utilizing proteins through the action of non-coding regulatory RNAs (sRNA). Notably, our previous research unveiled the significance of IsrR, a staphylococcal sRNA required for optimal growth in iron-depleted media and sustaining full virulence (Coronel-Tellez, 2022). Triggered by iron starvation, IsrR coordinates the down-regulation of genes encoding [Fe-S]-containing enzymes, including aconitase.
Aconitase converts citrate to isocitrate. CcpE, a citrate-activated transcriptional regulator, positively regulates its gene. In this new work, we show that IsrR inhibits the translation of aconitase mRNA and of ccpE mRNA, thus establishing a coherent feed-forward regulatory loop. This dual mechanism of repression ensures effective control of aconitase production by IsrR. Aconitase is a protein known for its second (moonlighting) activity in the absence of its [Fe-S] cluster; it becomes an RNA-binding protein (RBP) with regulatory capacity. This characteristic is conserved from prokaryotes to eukaryotes. Here, we show that the RBP activity of S. aureus aconitase reduces the amount of aconitase and influences the expression of genes encoding metabolic enzymes during iron deficiency.
Our study reveals the complex network of post-transcriptional regulations governing aconitase production that enables S. aureus to thrive in iron-deficient conditions. Importantly, these conditions are encountered by bacteria in the host as a result of a defense response known as nutritional immunity, in which iron and other essential nutrients are sequestered to prevent infection. More information: https://academic.oup.com/nar/advance-article/doi/10.1093/nar/gkae506/7692338 Contact: Philippe Bouloc philippe.bouloc@i2bc.paris-saclay.fr
|
|
Scooped by
I2BC Paris-Saclay
May 15, 5:27 AM
|
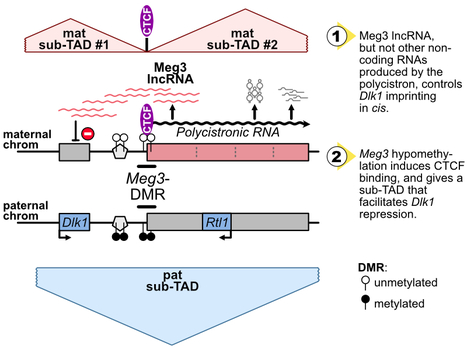
Imprinting gene regulation: Importance of a long non-coding RNA and of DNA methylation levels at an essential CTCF binding site
The Dlk1-Meg3-Dio3 domain is imprinted. Scientists at the IGMM and the I2BC have determined that Meg3 long non-coding RNA, as well as DNA methylation levels at an essential CTCF binding site in the domain, control Dlk1 imprinting in cis. The imprinted Dlk1-Dio3 domain comprises the Dlk1 and Rtl1 developmental genes that are solely expressed from the paternal chromosome, and a maternally-expressed polycistron that is further processed into the Meg3 long non-coding RNA (lncRNA) and many other small non-coding RNAs. In collaborative work between scientists at the IGMM (R. Feil’s group; Montpellier) and at the I2BC (D. Noordermeer’s group; Gif-sur-Yvette), researchers studied the precise role of Meg3 lncRNA and of its promoter for the control of Dlk1 and Rtl1 imprinted gene expression. Using mutant cells with either premature termination of transcription or deletion of part of the polycistron, they found that Meg3 lncRNA, but not other non-coding RNAs produced by the polycistron, controls Dlk1 imprinting in cis. The maternal expression of this polycistron is driven by the Meg3 differentially methylated region (Meg3-DMR) which is unmethylated on the maternal allele and acts there as an active promoter. Besides, the Meg3-DMR includes a known binding site for the architectural CTCF protein, that binds to the unmethylated maternal allele only. In this study, the researcher showed that the maternal Dlk1-Dio3 locus is organized into sub-Topologically Associating domains (sub-TADs) that are hinged by the allelic binding of CTCF to the maternal Meg3-DMR. They further found that the methylation levels at the Meg3-DMR are instructive for CTCF binding and thereby dictate distinctive sub-TAD organization at the two parental alleles which, in turns, facilitates Dlk1 repression on the maternal allele. Altogether, this collaborative work revealed that, the maternally-expressed Meg3 lncRNA controls Dlk1 silencing in cis, and that the low methylation levels at the maternal Meg3-DMR allows for specific sub-TADs structuration that concurrently contributes to Dlk1 silencing. More information: https://academic.oup.com/nar/advance-article/doi/10.1093/nar/gkae247/7645245?searchresult=1 Contact: Benoit Moindrot benoit.moindrot@i2bc.paris-saclay.fr
|
|
Scooped by
I2BC Paris-Saclay
April 15, 8:39 AM
|
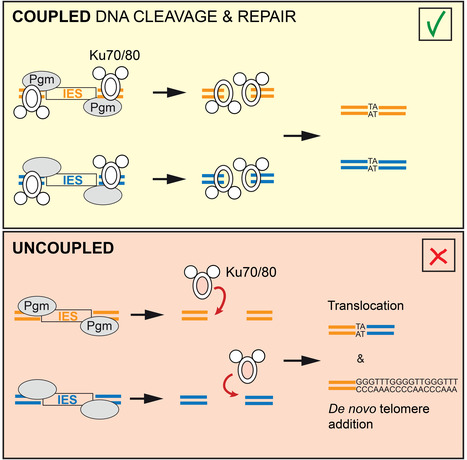
Uncoupling programmed DNA cleavage and repair scrambles the Paramecium somatic genome
No Ku – no cut: how the ciliate Paramecium tetraurelia protects its somatic genome from translocations and aberrant chromosome fragmentation during developmentally programmed rearrangements. DNA double-strand breaks (DSBs) are a major threat to genome integrity. When incorrectly repaired, they can lead to genome rearrangements, chromosome instability, diseases, or cell death. Yet, key physiological processes, such as antibody gene assembly or meiotic recombination, rely on DSB induction. How living organisms cope with programmed DSBs is a major question in genome biology. The “Programmed genome rearrangements” team studies Paramecium to dissect developmentally programmed DNA elimination mechanisms. During each sexual cycle, tens of thousands of germline Internal Eliminated Sequences (IESs), scattered all along chromosomes, undergo precise excision. This process requires DSB introduction at IES boundaries, followed by error-free stitching of flanking DNA fragments by the non-homologous end joining (NHEJ) pathway. The team previously reported that the presence of a specialized variant of the NHEJ factor Ku is a pre-requisite for the activation of DNA cleavage, indicating that the two steps of the reaction are coupled. In this study, they engineered a DNA binding-deficient Ku mutant, which they characterized in collaboration with the IntGen team from the B3S department. They showed that the mutant Ku activates DNA cleavage, but is defective in repair. Unrepaired broken ends at IES boundaries are then trimmed and healed by telomere addition. By co-expressing wild-type Ku with the mutant, they were able to uncouple the cleavage and repair steps during IES excision. Using high throughput DNA sequencing and novel dedicated bioinformatic tools, they showed that chromosome translocations were largely favored under these conditions. This work demonstrates that coupling DNA cleavage and DSB repair ensures faithful genome assembly during programmed rearrangements. More information: https://www.cell.com/cell-reports/fulltext/S2211-1247(24)00329-2 Contact: Mireille Bétermier mireille.betermier@i2bc.paris-saclay.fr – Julien Bischerour – julien.bischerour@i2bc.paris-saclay.fr
|
|
Scooped by
I2BC Paris-Saclay
March 25, 9:45 AM
|
Bettencourt-Schueller Foundation seminar on Art & Science: How does creativity emerge?
Chloé @Chl0e_Girard and Daan @DaanNoordermeer were invited to an interdisciplinary workshop organized by the Bettencourt-Schueller Foundation @Fondation_BS about creativity as motor for scientific research. A 3-days seminar was organized by the Bettencourt-Schueller Foundation at the Domaine de Chaumont-sur-Loire for former laureates (Prix Jeune Chercheur, Prix Impulsience, Prix Coups d’Elan pour la Recherche Française). This interdisciplinary seminar centered around creativity combined Arts & Science, the two main components of the foundation's patronage. The I2BC scientists participated in thematic workshops around Music, Mathematics, Visual Arts and Biology, presented by world-reknown artists and scientists. Daan Nordermeer was a recipient of the "Coup d'élan pour la recherche française”, while Chloé Girard received the "Prix jeune Chercheur", both in 2015.
|
|
Scooped by
I2BC Paris-Saclay
March 25, 6:10 AM
|
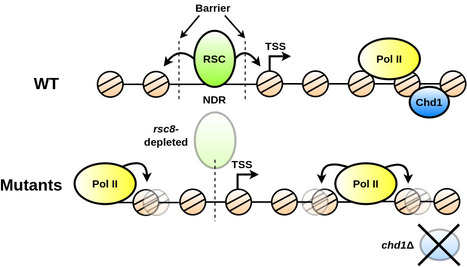
A genome-wide comprehensive analysis of nucleosome positioning in yeast
We show that the regulation and compartmentalisation of nucleosomal organisation require the concomitant actions of local and global processes that are maintained actively by energy consuming factors. In eukaryotic cells, the one-dimensional DNA molecules need to be tightly packaged into the spatially constraining nucleus. Folding is achieved on its lowest level by wrapping the DNA around nucleosomes. Their arrangement regulates other nuclear processes, such as tran- scription and DNA repair. Despite strong efforts to study nucleosome positioning using Next Generation Sequencing (NGS) data, the mechanism of their collective arrangement along the gene body remains poorly understood. Here, we classify nucleosome distributions of protein-coding genes in Saccharomyces cerevisiae according to their profile similarity and analyse their differences using functional Principal Component Analysis. By decomposing the NGS signals into their main descriptive functions, we compared wild type and chromatin remodeler-deficient strains, keeping position-specific details preserved whilst considering the nucleosome arrangement as a whole. A correlation analysis with other genomic proper- ties, such as gene size and length of the upstream Nucleosome Depleted Region (NDR), identified key factors that influence the nucleosome distribution. We reveal that the RSC chromatin remodeler—which is responsible for NDR maintenance—is indispensable for decoupling nucleosome arrangement within the gene from positioning outside, which inter- fere in rsc8-depleted conditions. Moreover, nucleosome profiles in chd1Δ strains displayed a clear correlation with RNA polymerase II presence, whereas wild type cells did not indicate a noticeable interdependence. We propose that RSC is pivotal for global nucleosome orga- nisation, whilst Chd1 plays a key role for maintaining local arrangement. More information: https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1011799 Contact: Arach GOLDAR <arach.goldar@i2bc.paris-saclay.fr>
|
|
Scooped by
I2BC Paris-Saclay
December 27, 2023 7:52 AM
|
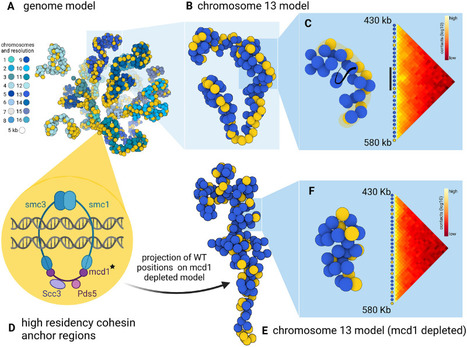
3D models of fungal chromosomes to enhance visual integration of omics data
It has become easier to build 3D models of the entire genome based on Hi-C data, and thus benefit from the methodology and visualization tools developed for structural biology. In this paper, we show how seeing the spatial organization of chromosomes can bring new perspectives to omics data integration. The functions of eukaryotic chromosomes and their spatial architecture in the nucleus are reciprocally dependent. Hi-C experiments are routinely used to study chromosome 3D organization by probing chromatin interactions. Standard representation of the data has relied on contact maps that show the frequency of interactions between parts of the genome. In parallel, it has become easier to build 3D models of the entire genome based on the same Hi-C data, and thus benefit from the methodology and visualization tools developed for structural biology. 3D modeling of entire genomes leverages the understanding of their spatial organization. However, this opportunity for original and insightful modeling is underexploited. In this paper, we show how seeing the spatial organization of chromosomes can bring new perspectives to omics data integration. We assembled state-of-the-art tools into a workflow that goes from Hi-C raw data to fully annotated 3D models and we re-analysed public omics datasets available for three fungal species. Besides the well-described properties of the spatial organization of their chromosomes (Rabl conformation, hypercoiling and chromosome territories), our results highlighted (i) in Saccharomyces cerevisiae, the backbones of the cohesin anchor regions, which were aligned all along the chromosomes, (ii) in Schizosaccharomyces pombe, the oscillations of the coiling of chromosome arms throughout the cell cycle and (iii) in Neurospora crassa, the massive relocalization of histone marks in mutants of heterochromatin regulators. 3D modeling of the chromosomes brings new opportunities for visual integration of omics data. This holistic perspective supports intuition and lays the foundation for building new concepts. More information: https://academic.oup.com/nargab/article/5/4/lqad104/7458894 Contact: Gaëlle LELANDAIS <gaelle.lelandais@i2bc.paris-saclay.fr>
|
|
Scooped by
I2BC Paris-Saclay
October 23, 2023 4:53 AM
|
Alicia Nevers: new assistant professor in the Genome Biology Department
Meet Alicia Nevers, an assistant professor of Paris-Saclay University, that just joined the Genome Biology Department of the I2BC. Alicia began her undergraduate studies with a DUT in Biological Engineering, which she obtained in 2011. She then went on to complete a Bachelor's degree in Life Sciences, followed by a Master's degree in Molecular and Cellular Biology, with a specialization in RNA Molecular Biology. From 2013 to 2017, she did her thesis with the Université Paris Sorbonne and in Alain Jacquier's laboratory at the Institut Pasteur. Under the supervision of Alain Jacquier and Gwenaël Badis-Bréard she studied, in the budding yeast Saccharomyces cerevisiae, the mechanisms of control of gene expression by transcriptional interference and the functionality of pervasive antisense transcription. Alicia then joined Mathieu Rougemaille's laboratory at I2BC as a research engineer between 2018 and 2020. She contributed to the discovery and characterization of a novel non-coding RNA involved in the control of meiosis in the fissiparous yeast Schizosaccharomyces pombe. After that, Alicia was hosted in Micalis Unit of Inrae as a post-doctoral researcher. Here, under the supervision of Didier Lereclus, Michel Gohar and Vincent Sanchis-Borja, she studied the role megaplasmids in new pathogens emergence. More particularly she studied pCER270 megaplasmid, representative of emetic strains of Bacillus cereus group. She particularly analyzed in natural and artificial hosts, the consequences of pCER270 presence for gene expression and biofilm formation ability. Now that she joined Frédéric Boccard’s laboratory as a senior lecturer, she’ll work under the supervision of Vicky Lioy and Stéphany Bury-Moné to describe a link between gene expression, chromosome conformation and bacterial lifestyle, in particular biofilm formation, in the pathogen Pseudomonas aeruginosa.
|
|
Scooped by
I2BC Paris-Saclay
September 19, 2023 10:52 AM
|
Dynamics of the Streptomyces chromosome: chance and necessity
Review in Trends in Genetics: Towards an integrated view of Streptomyces chromosome dynamics in space and time Streptomyces are prolific producers of specialized metabolites with applications in medicine and agriculture. Remarkably, these bacteria possess a large linear chromosome that is genetically compartmentalized: core genes are grouped in the central part, while the ends are populated by poorly conserved genes including antibiotic biosynthetic gene clusters. The genome is highly unstable and exhibits distinct evolutionary rates along the chromosome. Recent chromosome conformation capture (3C) and comparative genomics studies have shed new light on the interplay between genome dynamics in space and time. Here, we review insights that illustrate how the balance between chance (random genome variations) and necessity (structural and functional constraints) may have led to the emergence of spatial structuring of the Streptomyces chromosome. More information: https://www.cell.com/trends/genetics/fulltext/S0168-9525(23)00166-X Contact: Stéphanie BURY-MONE <stephanie.bury-mone@i2bc.paris-saclay.fr>
|
|
Scooped by
I2BC Paris-Saclay
July 19, 2023 10:24 AM
|
The formation of structural domains in chromosomes: a highly dynamic process to create stable regulatory functions
Our chromosomes are divided into strucrural domains that focus biological activity. In this literature review, researchers from the I2BC summarize recent insights into the dynamic nature of this process, and how this nonetheless can create stable regulation. Mammalian chromosomes are organized at different length scales within the cell nucleus. Topologically Associating Domains (TADs) are structural units of 3D genome organization that compartmentalize chromosomes into separated domains. Within these domains, biological functions are focused, thereby restricting the “spread” of gene regulation, DNA replication, recombination and repair. In this review, published in Current Opinion in Structural Biology, scientists from the Chromatin Dynamics team at the I2BC discuss recent insights into the structure and function of TADs. Whereas TADs were initially interpreted as insulated domains, recent studies are revealing that these domains should be interpreted as dynamic collections of actively extruding loops. This process of loop extrusion is subsequently blocked at dedicated TAD boundaries, thereby promoting intra-domain interactions over their surroundings. The authors discuss how mammalian TAD structure can emerge from this dynamic process and they discuss recent evidence that boundaries between TADs can have regulatory functions. More information: https://www.sciencedirect.com/science/article/abs/pii/S0959440X23000969 Contact: Daan Noordermeer <dann.noordermeer@i2bc.paris-saclay.fr>
|
|
Scooped by
I2BC Paris-Saclay
June 16, 2023 9:29 AM
|
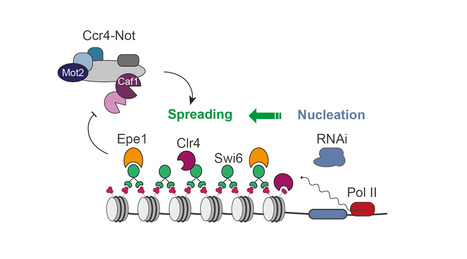
The Ccr4-Not complex is a major regulator of gene silencing and heterochromatin spreading
The fission yeast Ccr4-Not complex promotes propagation of the repressive H3K9me3 histone mark to mediate heterochromatic gene silencing. Eukaryotic genomes are partitioned into relaxed, gene-rich regions and condensed, gene-poor domains called heterochromatin. The maintenance of heterochromatin is crucial for proper genome expression and integrity, and requires multiple factors regulating histone modifications and/or the levels of RNA molecules produced from these regions. Such effectors not only promote heterochromatin assembly but also ensure its propagation from specific nucleation sites to defined domain boundaries. However, while the mechanisms involved in initiation of heterochromatin formation have been well documented, the molecular and biochemical properties underlying its spreading remain largely elusive. By combining genetic and single-cell approaches, we report here that the fission yeast Ccr4-Not complex, a multisubunit complex conserved throughout eukaryotes, is essential for efficient heterochromatin spreading to repress expression of nucleation-distal RNAs. The two catalytic activities of the complex, RNA deadenylation and protein ubiquitinylation, are each critical, thereby defining a dual enzymatic requirement in the process. More information: https://academic.oup.com/genetics/advance-article/doi/10.1093/genetics/iyad108/7190671?login=false Contact: Mathieu Rougemaille <mathieu.rougemaille@i2bc.paris-saclay.fr>
|
|
Scooped by
I2BC Paris-Saclay
May 17, 2023 11:50 AM
|
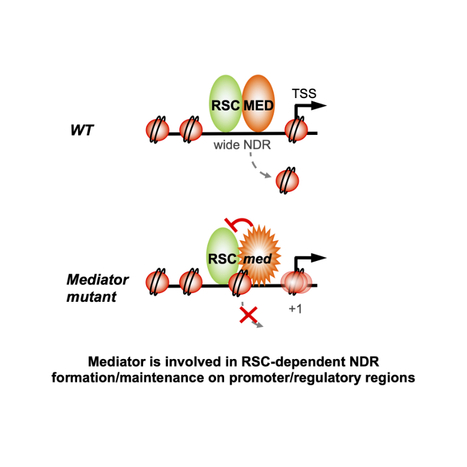
Two essential complexes--Mediator and RSC chromatin remodeler--work together for chromatin organization at promoters
Physical interaction and functional interplay between essential co-regulators in transcription and chromatin organization, Mediator and RSC chromatin remodeler, contributes to nucleosome-depleted region formation at promoters and +1 nucleosome positioning. Eukaryotic DNA is compactly structured through chromatin organization which governs and influences all DNA transactions. Regulation of transcription is a key phenomenon of gene expression whose alterations lead to severe human pathologies. Multisubunit coregulator complexes can act on chromatin structure as chromatin modifiers or remodelers, or stimulate the assembly of the transcriptional machinery. Mediator is an essential and conserved coactivator thought to act in concert with chromatin regulators. However, it remains largely unknown how their functions are coordinated. The team of J. Soutourina (Genome biology/I2BC and Institute Joliot, CEA/CNRS/Paris-Saclay) provides evidence in the budding yeast that Mediator establishes physical contact with RSC (Remodels the Structure of Chromatin), a conserved and essential chromatin remodeling complex that is crucial for nucleosome-depleted region (NDR) formation. The role of Mediator-RSC interaction in their chromatin binding, nucleosome occupancy and transcription was determined on a genomic scale. Mediator and RSC co-localize on wide NDRs of promoter regions, and specific Mediator mutations affect nucleosome eviction and stability of +1 nucleosome associated with transcription-start site. This work shows that Mediator contributes to RSC remodeling function to shape NDRs and maintain chromatin organization on promoter regions. It will help in our understanding of transcriptional regulation in the chromatin context relevant for severe diseases. More information: doi.org/10.1016/j.celrep.2023.112465 Contact: Julie Soutourina <julie.soutourina@i2bc.paris-saclay.fr>
|
|
Scooped by
I2BC Paris-Saclay
February 17, 2023 5:33 AM
|
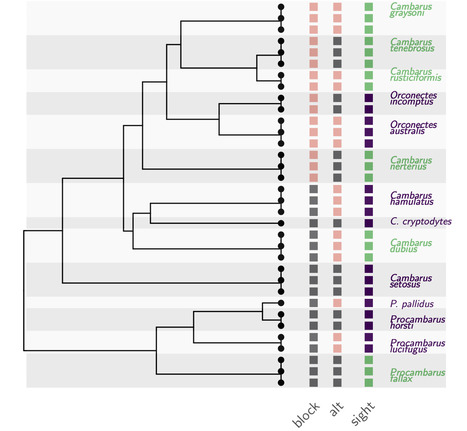
A Phylogenetic Framework to Simulate Synthetic Interspecies RNA-Seq Data
Understanding how to properly analyze your interspecies gene expression datasets. Interspecies RNA-Seq datasets are increasingly common, and have the potential to answer new questions about the evolution of gene expression. Single-species differential expression analysis is now a well-studied problem that benefits from sound statistical methods. Extensive reviews on biological or synthetic datasets have provided the community with a clear picture on the relative performances of the available methods in various settings. However, synthetic dataset simulation tools are still missing in the interspecies gene expression context. In this work, we develop and implement a new simulation framework. This tool builds on both the RNA-Seq and the phylogenetic comparative methods literatures to generate realistic count datasets, while taking into account the phylogenetic relationships between the samples. We illustrate the usefulness of this new framework through a targeted simulation study, that reproduces the features of a recently published dataset, containing gene expression data in adult eye tissue across blind and sighted freshwater crayfish species. Using our simulated datasets, we perform a fair comparison of several approaches used for differential expression analysis. This benchmark reveals some of the strengths and weaknesses of both the classical and phylogenetic approaches for interspecies differential expression analysis, and allows for a reanalysis of the crayfish dataset. The tool has been integrated in the R package compcodeR, freely available on Bioconductor More information: https://academic.oup.com/mbe/article/40/1/msac269/6889356 Contact: Melina Gallopin <melina.gallopin@i2bc.paris-saclay.fr>
|
|
|
Scooped by
I2BC Paris-Saclay
July 26, 8:18 AM
|
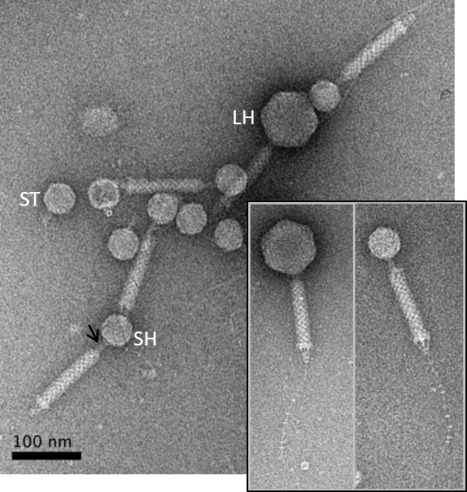
Acinetobacter baumannii satellite phage Aci01-2-Phanie depends on a helper myophage for its multiplication
Phanie is a phi-29 like phage that protects its genome inside non-infectious podovirus-like particles, requiring acquisition of the tail from a myovirus helper for production of infectious chimeras. Viruses of bacteria (bacteriophages or phages) display a remarkable genomic diversity reflecting the complex interaction they have built with their host during evolution. In addition to fully infectious viral particles, different types of genetic elements were described to require a helper phage for production of infectious virions. The genome of these satellite parasite sequences may share sequences with phages or be more closely related to non-viral chromosome islands. We describe a new mode of satellite phage dependence on a helper phage. Phanie, like the genetically related Bacillus subtilis phage phi29, replicates its linear dsDNA by a protein primed-mechanism and protects it inside podovirus-like particles. However, these particles are defective, requiring acquisition of the tail from a myovirus helper for production of infectious virions. Formation of chimeras between a phi29-like podovirus and a helper contractile tail reveals an unexpected association between very different bacterial viruses. More information: https://journals-asm-org.insb.bib.cnrs.fr/doi/10.1128/jvi.00667-24 Contact: Christine Pourcel christine.pourcel@i2bc.paris-saclay.fr
|
|
Scooped by
I2BC Paris-Saclay
July 1, 8:55 AM
|
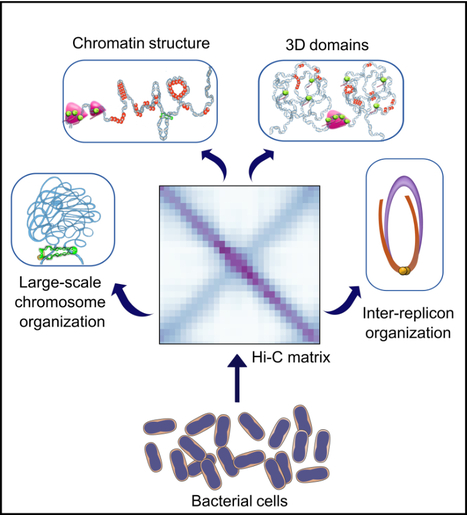
3D bacterial genome organization
In this work, we reviewed the latest Hi-C findings and discuss the principles of bacterial chromosome folding that can be inferred from them. The use of Hi-C not only revealed the diversity of 3D-genome architectures in multiple bacterial species across the phylogenetic tree, but also the different scales of genome organization and the mechanisms of action of key structural factors and processes. Here, we revisited the work performed on these bacteria and discussed the rules of genome folding that can be deduced from it. more information: https://onlinelibrary.wiley.com/doi/10.1111/mmi.15290 contact: Virginia LIOY <virginia.lioy@i2bc.paris-saclay.fr>
|
|
Scooped by
I2BC Paris-Saclay
April 15, 9:00 AM
|
How does DNA methylation, an epigenetic modification involved in cancer and disease, change the folding of our chromosomes?
In this literature review in Nature Structural and Molecular Biology, scientists from the Institute Jacques Monod and the I2BC discuss how the methylation of DNA changes the folding of our chromosomes and how this can cause defects and disease. DNA methylation is used as an epigenetic mark on human chromosomes to regulate the transcriptional activity of genes and parasitic transposable elements. Within the nucleus of human cells, chromosomes are non-randomly organized, which strongly influences transcriptional activity as well. DNA methylation and 3D chromosome organization are strongly reorganized during the earliest stages of embryonic development and in diseases like cancer. In this literature review, published in Nature Structural and Molecular Biology, scientists from the Institute Jacques Monod and the I2BC discuss the links between the two levels of chromosome organization. The authors particularly focus on the CTCF insulator protein, whose binding to the DNA directly translates 3D chromosome organization to transcriptional activity. CTCF binding to the DNA has been reported as DNA methylation sensitive in certain contexts, which for certain genes explains the link between transcriptional activity, 3D chromosome organization and DNA methylation. Yet, changes in CTCF binding near other genes appears less obviously coupled to this epigenetic mark, indicating that other mechanisms must play a role as well. more information: https://www.nature.com/articles/s41594-024-01241-6 contact: Daan Noordermeer <daan.noordermeer@i2bc.paris-saclay.fr>
|
|
Scooped by
I2BC Paris-Saclay
April 15, 8:37 AM
|
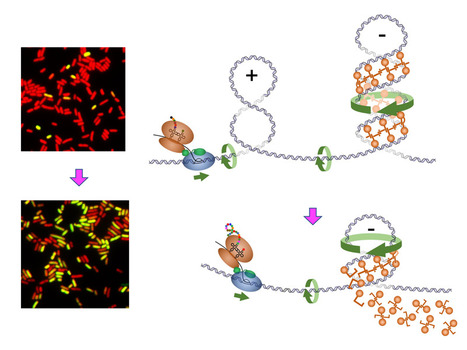
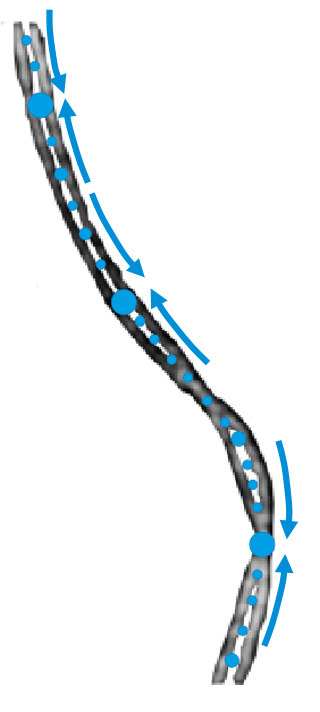
Transcription-induced DNA supercoiling clears RNA polymerase’s path in bacterial chromatin
Positive DNA supercoiling generated ahead of RNA polymerase during transcription elongation dislodges nucleoid-structuring protein H-NS from the DNA. Bacterial chromosomal DNA is condensed more than 1000 times inside the cell. This condensation results from a slight underwinding of the double helix, leading to the formation of loops of negatively supercoiled DNA (plectonemes) and from interactions with structural proteins that help compact these loops. However, these chromatin-associated proteins obstruct transcription and need to be transiently removed to allow gene expression. This is the case for H-NS, an abundant protein that blocks the expression of numerous genes. In Gram-negative pathogenic bacteria such as Salmonella, H-NS represses the expression of the majority of virulence genes. Displacement of H-NS becomes necessary during the infection process, which relies on the function of virulence gene products.
In a study published in the journal Nature Communications, researchers from the Genome Biology department of the Institute for Integrative Biology of the Cell (I2BC) present evidence that binding of H-NS to DNA is destabilized, from a distance, by the positive supercoils forming ahead of approaching RNA polymerase. It is known that the process of transcription induces DNA supercoiling by forcing the rotation of the DNA axis during elongation. To explain the destabilization of H-NS:DNA complexes by positive supercoiling, the researchers propose a model in which H-NS oligomers play a scaffolding role in negatively supercoiled DNA by bridging opposite arms of the plectonemic structure. Accumulation of positive supercoils and concomitant DNA axis rotation ahead of the approaching RNA polymerase cause the H-NS-bound, negatively supercoiled plectoneme to unroll, leading to the collapse of the scaffold and allowing RNA polymerase to continue on its path. More information: https://www.nature.com/articles/s41467-024-47114-w Contact: Lionelo Bossi lionello.bossi@i2bc.paris-saclay.fr
|
|
Scooped by
I2BC Paris-Saclay
March 25, 6:14 AM
|
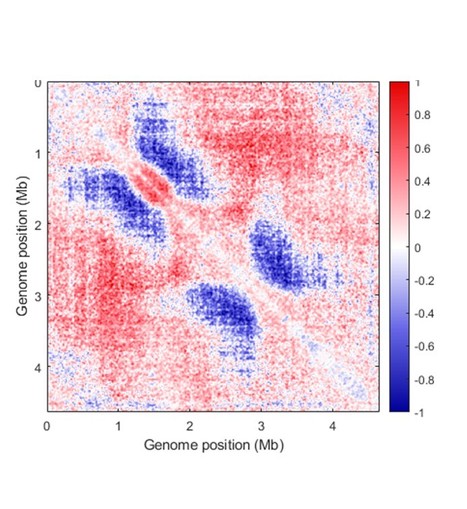
Activity of MukBEF for chromosome management in E. coli and its inhibition by MatP
The MukBEF condensin of E. coli loads at the replication fork and is unloaded at the Ter region by MatP. Structural maintenance of chromosomes (SMC) complexes share conserved structures and serve a common role in maintaining chromosome architecture. In the bacterium Escherichia coli, the SMC complex MukBEF is necessary for rapid growth and the accurate segregation and positioning of the chromosome, although the specific molecular mechanisms involved are still unknown. Here, we used a number of in vivo assays to reveal how MukBEF controls chromosome conformation and how the MatP/matS system prevents MukBEF activity. Our results indicate that the loading of MukBEF occurs preferentially on newly replicated DNA, at multiple loci on the chromosome where it can promote long-range contacts in cis even though MukBEF can promote long-range contacts in the absence of replication. Using Hi-C and ChIP-seq analyses in strains with rearranged chromosomes, the prevention of MukBEF activity increases with the number of matS sites and this effect likely results from the unloading of MukBEF by MatP. Altogether, our results reveal how MukBEF operates to control chromosome folding and segregation in E. coli. More information: https://elifesciences.org/articles/91185 Contact: Stéphane DUIGOU <stephane.duigou@i2bc.paris-saclay.fr>
|
|
Scooped by
I2BC Paris-Saclay
December 27, 2023 7:55 AM
|
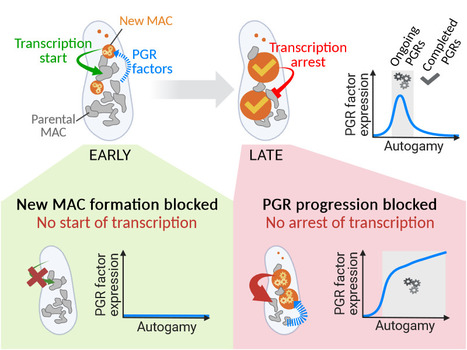
Inter-generational nuclear crosstalk controls the progression of the Paramecium developmental program
Perturbing the Paramecium transcriptome during somatic development reveals the existence of internuclear feed-forward and feedback transcriptional regulatory loops in a multinucleate unicellular eukaryote. The single-celled ciliate Paramecium tetraurelia harbors multiple nuclei within its cytoplasm: two germline micronuclei (MIC) dedicated to sexual reproduction and a somatic macronucleus (MAC) responsible for gene expression. During the sexual cycle, the parental MAC undergoes progressive degradation while, in the same cell, new MACs develop from copies of the zygotic MIC. Throughout this process, gene expression primarily occurs in the parental MAC, and clusters of developmentally regulated genes are successively switched on and off. Concomitantly, the genome of the new MACs is extensively rearranged and cleansed of its transposons and other DNA repeats, to become competent for gene expression. New MAC development has long been known to be controled by the parental MAC, which expresses key genes involved in programmed genome rearrangement (PGR). In this study, RNA deep sequencing and bioinformatic analysis of differential gene expression reveal a reciprocal relationship between the two generations of MACs: the formation of the new MACs is essential for the proper induction of a subset of developmental genes. Later in the sexual cycle, if PGR is hindered in the new MACs, a group of genes partially overlapping the former subset is upregulated, and no longer switched off. Thus, the progression of new MAC development regulates the gene expression program in the parental MAC at different stages. The set of co-expressed genes identified during the course of this work establishes a list of potential novel actors, whose roles in PGR will be explored in future studies. More information: https://doi.org/10.1093/nar/gkad1006 Contact: Olivier Arnaiz – Mireille Bétermier <olivier.arnaiz@i2bc.paris-saclay.fr> <mireille.betermier@i2bc.paris-saclay.fr>
|
|
Scooped by
I2BC Paris-Saclay
October 23, 2023 5:12 AM
|
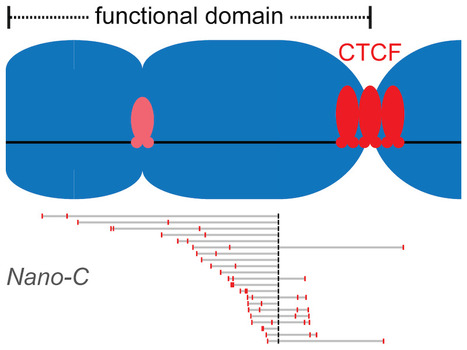
Clustered binding of the CTCF protein creates functional domains in the human and mouse genome with improved structure.
Clusters of binding sites for the CTCF protein create boundaries between functional domains in the mouse and human genomes. Scientists at the I2BC have determined the characteristics of these clusters and how individual sites contribute to domain separation. Topologically Associating Domains (TADs) regionalize the human and mouse genome into domains that are essential for processes like gene regulation and DNA repair. Most boundaries between TADs bind the CTCF insulator protein, which is responsible for the blocking of interactions between neighboring TADs. CTCF is thereby directly responsible for improved functional precision and reduced regulatory interference between domains in the genome. Although a clustering of CTCF binding sites had previously been observed at TAD boundaries, its structural impact remained to be determined.
Scientists in the Chromatin Dynamics group at the I2BC, in collaboration with groups at the Ecole Normale Supérieure (Paris) and the University of Pennsylvania (USA), found that three features of CTCF binding are enriched at TAD boundaries, including the presence of closely-spaced copies of its DNA binding motifs, a reduced distance between clusters of motifs and an enrichment of clusters with improved binding affinity. Using Nano-C technology, a new approach to measure the blocking of DNA interactions, they subsequently determined the blocking capacity of individual sites of CTCF binding. These studies reveal that these sites individually contribute to the blocking of interactions between domains, but in an incomplete manner. Grouping of multiple sites of CTCF binding in the genome thus creates a stepwise insulation between neighboring TADs, thereby improving the focus of genomic functions. More information: https://doi.org/10.1038/s41467-023-41265-y Contact: Daan NOORDERMEER <daan.noordermeer@i2bc.paris-saclay.fr>
|
|
Scooped by
I2BC Paris-Saclay
September 20, 2023 8:58 AM
|
ERC Starting Grant, Congratulations to Chloé Girard
Congratulations to Chloé Girard from the Meiotic Recombination and Pairing team, who has been awarded a ERC Starting Grant for her innovative project, DYNACO. This project focuses on the phenomenon of crossover interference during meiosis, a widely conserved mechanism first observed more than a century ago. The DYNACO project will advance our understanding of the genetic mechanisms underlying meiotic crossover formation and interference. It also offers potential applications for the manipulation of genetic recombination in plant breeding.
|
|
Scooped by
I2BC Paris-Saclay
September 19, 2023 10:40 AM
|
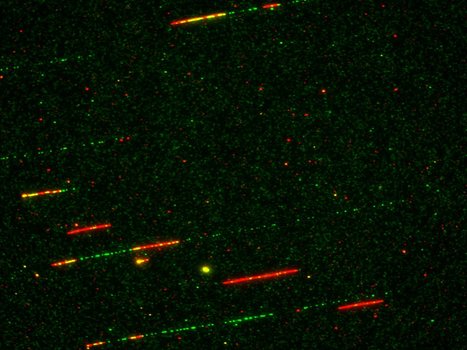
How Rif1 bridles rapid genome replication during early developmental stages in vertebrae
Combined experimental and in-sillico approaches show that the Rif1 protein restricts the activation of replication origins of chromatin domains and the recruitment of initiation factors during the first embryonic divisions. In multicellular eukaryotic organisms, genomic DNA replicates in a time-controlled manner by ordering early, intermediate, or late replication regions according to a spatio-temporal replication program. How this program is orchestrated is poorly understood, but its dysregulation leads to genomic instability often observed in cancer. The Rif1 protein is a key regulator of this program in eukaryotes; however, its role during the first embryonic cell cycles, when DNA replication is very rapid and replication factors are abundant, remained poorly characterized. Researchers from I2BC, NeuroPSI, ENS Paris, and CalTech (USA) have clarified these mechanisms using the high-performance in vitro system of Xenopus egg extracts by combining the analysis of DNA fibers by molecular combing with an in-sillico model. This study, published in Communications Biology, reveals that in the absence of Rif1, the temporal program of replication is greatly accelerated at the level of groups of origins. This acceleration is accompanied by increased chromatin recruitment of an S phase kinase (Cdc7/Drf1) and several other key factors implicated in the replication initiation (Treslin/MTBP, RecQL4). The model proposed in this study is that Rif1 simultaneously restricts access to DNA or the activity of several factors to fine-tune the exceptionally rapid DNA synthesis observed during embryonic development. A better understanding of the role of Rif1 during these stages opens a way to elucidating the molecular mechanisms involved in certain diseases resulting from Rif1 mutations or variants in humans.
Read on to find out more in Nature Portfolio: https://cellmolbiocommunity.springernature.com/posts/how-rif1-bridles-rapid-embryonic-dna-synthesis Contact: Kathrin MARHEINEKE <kathrin.marheineke@i2bc.paris-saclay.fr>
Portrait Jeune Chercheur - Frédéric Frottin, chercheur en biochimie des protéines
Chargé de recherche au CNRS depuis 2020, Frédéric Frottin est biochimiste cellulaire. Ses travaux de recherche se déroulent à l'I2BC (Institut de Biologie Intégrative de la Cellule, UMR 9198 CEA/CNRS/UPSaclay, Gif-sur-Yvette) au sein de l’équipe Maturation, Destinée cellulaire des protéines et Thérapeutiques. Frédéric s’intéresse aux mécanismes cellulaires assurant l’homéostasie des protéines et leur contrôle qualité. En particulier, il étudie d’une part le rôle des modifications protéiques sur ces aspects ainsi que celui des organites sans membrane. Ses travaux ont des implications pour de nombreuses maladies notamment des maladies neurodégénératives telles que les maladies d’Alzheimer et de Parkinson. Frédéric a obtenu son doctorat en 2011 après un travail qui déjà portait sur les modifications protéiques. En particulier, il a amélioré notre compréhension des bases moléculaires de l’excision de la méthionine N-terminale, qui est un mécanisme essentiel à la survie de tout organismes. De plus, il a révélé son rôle dans le maintien de l’homéostasie du glutathion et des protéines. Ces travaux ont été réalisé avec de nombreux organismes incluant la plante modèle Arabidopsis thaliana, des cellules humaines cultivées, la levure ainsi que des archéobactéries. Ayant développé un intérêt pour l’homéostasie des protéines durant sa thèse, fin 2011 Frédéric rejoint le département du Prof. F.U. Hartl à l’Institut Max Planck de biochimie à Munich. Durant son postdoc, il a étudié les mécanismes de secours qui sont engagés lorsque le protéome subit des dommages. En 2012, il obtient une bourse EMBO pour mener ses recherches. Notamment, il a travaillé sur des voies cellulaires importantes pour le contrôle de l’homéostasie des protéines qui aide à notre compréhension des mécanismes associés aux maladies neurodégénératives. Il a participé à l’amélioration de notre compréhension de la toxicité induite par les agrégats. Ces agrégats, qui sont des espèces protéiques aberrantes, sont retrouvés dans ces maladies mais aussi dans certains cancers. Ses travaux ont montré que les agrégats endommageaient des fonctions essentielles telles que la dégradation, la synthèse, et le transport des protéines. De plus, il a découvert un nouveau mécanisme de contrôle qualité des protéines dans le noyau cellulaire. Ce mécanisme est l’accumulation transitoire d’espèces protéiques aberrantes au sein du nucléole lors d’un stress. Le nucléole est un sous compartiment du noyau. Ce compartiment est sans membrane et de type liquide. Dans le nucléole, ces protéines aberrantes sont moins toxiques et ne forment pas d’agrégats irréversibles. En 2020, il est recruté au CNRS pour poursuivre ces recherches sur cette voie nouvellement découverte. “If you are not part of the solution, you are part of the precipitate” – Chemistry joke, attribué à Scott Trahan. Contact : frederic.frottin@i2bc.paris-saclay.fr
Via Life Sciences UPSaclay
|
|
Scooped by
I2BC Paris-Saclay
May 17, 2023 11:59 AM
|
A "coarse" solution to the mystery of crossover interference?
For over a century, the mystery of crossover interference has been puzzling: how do meiotic crossovers communicate with each others to establish their unique distribution along chromosomes? In this review, we discuss a new model that could explain it: the coarsening model. Meiotic crossovers, which are exchanges of genetic material between homologous chromosomes, are more evenly and distantly spaced along chromosomes than expected by chance. This is because the occurrence of one crossover reduces the likelihood of nearby crossover events – a conserved and intriguing phenomenon called crossover interference. Although crossover interference was first described over a century ago, the mechanism allowing coordination of the fate of potential crossover sites half a chromosome away remains elusive. In this review, we discuss the recently published evidence supporting a new model for crossover patterning, coined the coarsening model, and point out the missing pieces that are still needed to complete this fascinating puzzle. More information: here Contact: Chloé Girard <chloe.girard@i2bc.paris-saclay.fr>
|
|
Scooped by
I2BC Paris-Saclay
February 17, 2023 5:46 AM
|
Recipient chromosome-encoded UvrD helicase plays a role in plasmid acquisition by conjugation
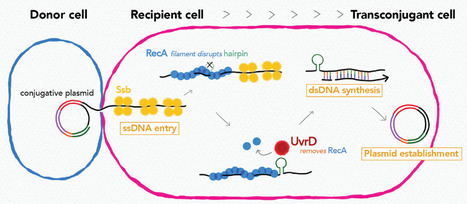
Research on conjugation unveiled that recipient chromosome-encoded UvrD helicase plays role in plasmid acquisition, shedding light on important DNA actions in the early stage of transconjugant cell establishment. Conjugative plasmids are a major dissemination source of resistance to antibiotics in bacterial populations. Even though they encode genes necessary and sufficient for their horizontal transmission, other environmental and genetic factors can alter the efficiency of plasmid transmission. In this study by using Transposon-insertion site sequencing and genetics experiments, we showed that defects in a conserved DNA helicase, UvrD impairs transfer of various conjugative plasmids. This was observed specifically in the recipient cell, not in the donor cell. During conjugation, one strand of plasmid DNA transfers from the donor to the recipient cell. By time-lapse microscopy with sophisticated monitoring tools to detect single-stranded (ss) and double-stranded (ds)DNA, we showed that UvrD plays a role in successful ss to ds conversion of the acquired DNA. An interesting spin is that UvrD homologs are well-known to be multifunctional and many of them are involved in different DNA repair pathways. Our work showed that in addition to its role in maintaining genome integrity, UvrD is also key for the establishment of horizontally acquired DNA that drives genome diversity and evolution. More information: https://doi.org/10.1093/nar/gkad075 Contact: Yoshiharu Yamaichi <yoshiharu.yamaichi@i2bc.paris-saclay.fr>
|