Your new post is loading...
Your new post is loading...

|
Scooped by
Gerd Moe-Behrens
November 4, 2025 8:25 AM
|
Cooperative catalysis, in which multiple catalytic units operate synergistically, underpins a variety of synthetically and mechanistically important organic reactions1–4. Despite its potential utility in new reactivity contexts, approaches to the discovery of cooperative catalysts have been limited, typically relying on serendipity or on prior knowledge of single-catalyst reactivity1,5. Systematic searches for unanticipated types of catalyst cooperativity must contend with vast combinatorial complexity and are therefore not undertaken6–10. Here, we describe a pooling–deconvolution algorithm, inspired by group testing11, that identifies cooperative catalyst behaviors with low experimental cost while accommodating potential inhibitory effects between catalyst candidates. The workflow was validated first on simulated cooperativity data, and then by experimentally identifying previously documented cooperativity between organocatalysts in an enantioselective oxetane-opening reaction. The workflow was then applied in a discovery context to a Pd-catalyzed decarbonylative cross-coupling reaction, enabling the identification of several ligand pairs that promote the target transformation at substantially lower catalyst loading and temperature than previously reported with single ligand systems.
|
|
Scooped by
Gerd Moe-Behrens
September 19, 2025 10:21 AM
|
Scientists used AI to write coherent viral genomes, using them to synthesize bacteriophages capable of killing resistant strains of bacteria.
|
|
Scooped by
Gerd Moe-Behrens
July 13, 2025 10:41 AM
|
Multicellular coordination enhances biological complexity, yet the widely used yeast Saccharomyces cerevisiae possesses limited multicellular capabilities. Here, we expand the possibilities for engineering multicellular behaviors in yeast by developing modular toolkits for two key mechanisms in multicellularity, contact-dependent signaling and specific cell-cell adhesion. MARS (mating-peptide anchored response system) enables contact-dependent signaling via surface-displayed peptides and G protein-coupled receptors, mimicking juxtacrine communication, while Saccharomyces SATURN (adhesion toolkit for multicellular patterning) uses adhesion-protein pairs for the creation of programmable cell aggregation patterns. Combining these allows the construction of multicellular logic circuits, equivalent to developmental programs that lead to cell differentiation based on local population. We further created JUPITER (juxtacrine sensor for protein-protein interaction), a genetic sensor based on MARS and SATURN, for assaying protein-protein interactions and selecting high-affinity nanobody binders. Collectively, these toolkits present versatile building blocks for constructing complex, user-defined multicellular yeast systems and expand the scope of its biotechnological applications.
|
|
Scooped by
Gerd Moe-Behrens
June 2, 2025 7:59 AM
|
Global energy production can't keep pace with AI's demands
|
|
Scooped by
Gerd Moe-Behrens
May 19, 2025 10:00 AM
|
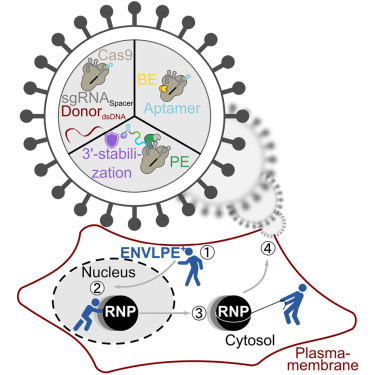
Engineered nucleocytosolic vehicles for loading of programmable editors
Advanced gene editing methods have accelerated biomedical discovery and hold great therapeutic promise, but safe and efficient delivery of gene editors remains challenging. In this study, we present a virus-like particle (VLP) system featuring nucleocytosolic shuttling vehicles that retrieve pre-assembled Cas-effectors via aptamer-tagged guide RNAs. This approach ensures preferential loading of fully assembled editor ribonucleoproteins (RNPs) and enhances the efficacy of prime editing, base editing, trans-activators, and nuclease activity coupled to homology-directed repair in multiple immortalized, primary, stem cell, and stem-cell-derived cell types. We also achieve additional protection of inherently unstable prime editing guide RNAs (pegRNAs) by shielding the 3′-exposed end with Csy4/Cas6f, further enhancing editing performance. Furthermore, we identify a minimal set of packaging and budding modules that can serve as a platform for bottom-up engineering of enveloped delivery vehicles. Notably, our system demonstrates superior per-VLP editing efficiency in primary T lymphocytes and two mouse models of inherited retinal disease, highlighting its therapeutic potential.
|
|
Scooped by
Gerd Moe-Behrens
January 16, 2025 8:07 AM
|
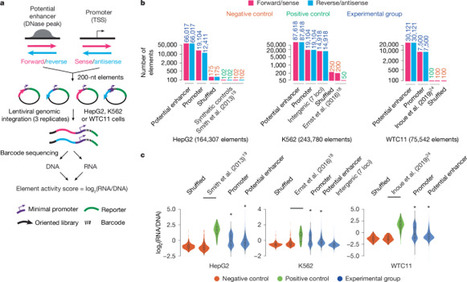
The human genome contains millions of candidate cis-regulatory elements (cCREs) with cell-type-specific activities that shape both health and many disease states1. However, we lack a functional understanding of the sequence features that control the activity and cell-type-specific features of these cCREs. Here we used lentivirus-based massively parallel reporter assays (lentiMPRAs) to test the regulatory activity of more than 680,000 sequences, representing an extensive set of annotated cCREs among three cell types (HepG2, K562 and WTC11), and found that 41.7% of these sequences were active. By testing sequences in both orientations, we find promoters to have strand-orientation biases and their 200-nucleotide cores to function as non-cell-type-specific ‘on switches’ that provide similar expression levels to their associated gene. By contrast, enhancers have weaker orientation biases, but increased tissue-specific characteristics. Utilizing our lentiMPRA data, we develop sequence-based models to predict cCRE function and variant effects with high accuracy, delineate regulatory motifs and model their combinatorial effects. Testing a lentiMPRA library encompassing 60,000 cCREs in all three cell types further identified factors that determine cell-type specificity. Collectively, our work provides an extensive catalogue of functional CREs in three widely used cell lines and showcases how large-scale functional measurements can be used to dissect regulatory grammar. Lentivirus-based reporter assays for 680,000 regulatory sequences from three cell lines coupled to machine-learning models lead to insights into the grammar of cis-regulatory elements.
|
|
Scooped by
Gerd Moe-Behrens
January 16, 2025 8:06 AM
|
Emerging protein-design competitions aim to sift out the functional from the fantastical. But researchers hope that the real prize will be a revolution for the field.
|
|
Scooped by
Gerd Moe-Behrens
December 12, 2024 12:01 PM
|
Non-ribosomal peptide synthetases are assembly line biosynthetic pathways that are used to produce critical therapeutic drugs and are typically arranged as large multi-domain proteins called megasynthetases1. They synthesize polypeptides using peptidyl carrier proteins that shuttle each amino acid through modular loading, modification and elongation2 steps, and remain challenging to structurally characterize, owing in part to the inherent dynamics of their multi-domain and multi-modular architectures3. Here we have developed site-selective crosslinking probes to conformationally constrain and resolve the interactions between carrier proteins and their partner enzymatic domains4,5. We apply tetrazine click chemistry to trap the condensation of two carrier protein substrates within the active site of the condensation domain that unites the first two modules of tyrocidine biosynthesis and report the high-resolution cryo-EM structure of this complex. Together with the X-ray crystal structure of the first carrier protein crosslinked to its epimerization domain, these structures highlight captured intermodular recognition events and define the processive movement of a carrier protein from one catalytic step to the next. Characterization of these structural relationships remains central to understanding the molecular details of these unique synthetases and critically informs future synthetic biology design of these pathways. Structural studies of tyrocidine synthetase using site-selective crosslinking probes to link condensation domains with carrier protein substrates define key interactions and molecular mechanisms of non-ribosomal protein synthesis.
|
|
Scooped by
Gerd Moe-Behrens
October 25, 2024 7:31 PM
|
Cis-regulatory elements (CREs) control gene expression, orchestrating tissue identity, developmental timing and stimulus responses, which collectively define the thousands of unique cell types in the body1–3. While there is great potential for strategically incorporating CREs in therapeutic or biotechnology applications that require tissue specificity, there is no guarantee that an optimal CRE for these intended purposes has arisen naturally. Here we present a platform to engineer and validate synthetic CREs capable of driving gene expression with programmed cell-type specificity. We take advantage of innovations in deep neural network modelling of CRE activity across three cell types, efficient in silico optimization and massively parallel reporter assays to design and empirically test thousands of CREs4–8. Through large-scale in vitro validation, we show that synthetic sequences are more effective at driving cell-type-specific expression in three cell lines compared with natural sequences from the human genome and achieve specificity in analogous tissues when tested in vivo. Synthetic sequences exhibit distinct motif vocabulary associated with activity in the on-target cell type and a simultaneous reduction in the activity of off-target cells. Together, we provide a generalizable framework to prospectively engineer CREs from massively parallel reporter assay models and demonstrate the required literacy to write fit-for-purpose regulatory code. A generalizable framework to prospectively engineer cis-regulatory elements from massively parallel reporter assay models can be used to write fit-for-purpose regulatory code.
|
|
Scooped by
Gerd Moe-Behrens
July 22, 2024 6:10 PM
|
For Lucas Farnung, there is no question more fascinating than how a single fertilized egg develops into a fully-functioning human. As a structural biologist, he is studying this process on the smallest scale: the trillions of atoms that must synchronize their work to make it happen.
|
|
Scooped by
Gerd Moe-Behrens
July 12, 2024 7:51 AM
|
Researchers analysed thousands of laboratory-made plasmids and discovered that nearly half of them had defects, raising questions of experimental reproducibility.
|
|
Scooped by
Gerd Moe-Behrens
July 3, 2024 10:08 AM
|
Single-cell and spatial molecular profiling assays have shown large gains in sensitivity, resolution and throughput. Applying these technologies to specimens from human and model organisms promises to comprehensively catalogue cell types, reveal their lineage origins in development and discern their contributions to disease pathogenesis. Moreover, rapidly dropping costs have made well-controlled perturbation experiments and cohort studies widely accessible, illuminating mechanisms that give rise to phenotypes at the scale of the cell, the tissue and the whole organism. Interpreting the coming flood of single-cell data, much of which will be spatially resolved, will place a tremendous burden on existing computational pipelines. However, statistical concepts, models, tools and algorithms can be repurposed to solve problems now arising in genetic and molecular biology studies of development and disease. Here, I review how the questions that recent technological innovations promise to answer can be addressed by the major classes of statistical tools. Single-cell, spatial and multi-omic profiling technologies generate large-scale data that reveal the output of genome-scale experiments across diverse cells, tissues and organisms. Cole Trapnell reviews the underlying core statistical challenges that need to be tackled to harness the power of these technologies and advance our understanding of gene function in health and disease.
|
|
Scooped by
Gerd Moe-Behrens
April 11, 2024 2:32 PM
|
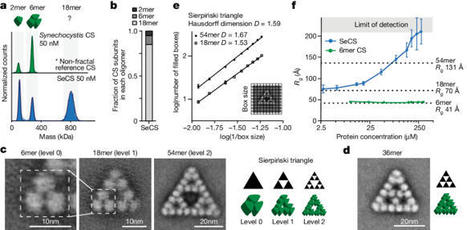
Fractals are patterns that are self-similar across multiple length-scales1. Macroscopic fractals are common in nature2–4; however, so far, molecular assembly into fractals is restricted to synthetic systems5–12. Here we report the discovery of a natural protein, citrate synthase from the cyanobacterium Synechococcus elongatus, which self-assembles into Sierpiński triangles. Using cryo-electron microscopy, we reveal how the fractal assembles from a hexameric building block. Although different stimuli modulate the formation of fractal complexes and these complexes can regulate the enzymatic activity of citrate synthase in vitro, the fractal may not serve a physiological function in vivo. We use ancestral sequence reconstruction to retrace how the citrate synthase fractal evolved from non-fractal precursors, and the results suggest it may have emerged as a harmless evolutionary accident. Our findings expand the space of possible protein complexes and demonstrate that intricate and regulatable assemblies can evolve in a single substitution. Citrate synthase from the cyanobacterium Synechococcus elongatus is shown to self-assemble into Sierpiński triangles, a finding that opens up the possibility that other naturally occurring molecular-scale fractals exist.
|
|
|
Scooped by
Gerd Moe-Behrens
October 3, 2025 7:12 PM
|
Ordering DNA for AI-designed toxins doesn’t always raise red flags.
|
|
Scooped by
Gerd Moe-Behrens
September 16, 2025 8:14 AM
|
Amid growing debates about the benefits and risks of studying looking-glass versions of life’s building blocks, there is an urgent need to bridge divergent views.
|
|
Scooped by
Gerd Moe-Behrens
July 13, 2025 10:39 AM
|
|
|
Scooped by
Gerd Moe-Behrens
May 23, 2025 7:06 AM
|
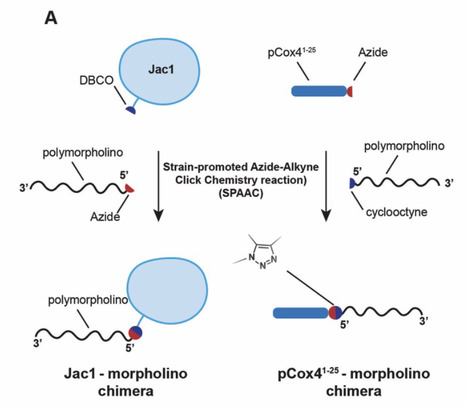
Mitochondria fulfill central functions in metabolism and energy supply. They express their own genome, which encodes key subunits of the oxidative phosphorylation system. However, central mechanisms underlying mitochondrial gene expression remain enigmatic. A lack of suitable technologies to target mitochondrial protein synthesis in cells has limited experimental access. Here, we silenced the translation of specific mitochondrial mRNAs in living human cells by delivering synthetic peptide-morpholino chimeras. This approach allowed us to perform a comprehensive temporal monitoring of cellular responses. Our study provides insights into mitochondrial translation, its integration into cellular physiology, and provides a strategy to address mitochondrial gene expression in living cells. The approach can potentially be used to analyze mechanisms and pathophysiology of mitochondrial gene expression in a range of cellular model systems.
|
|
Scooped by
Gerd Moe-Behrens
January 21, 2025 4:27 PM
|
|
|
Scooped by
Gerd Moe-Behrens
January 16, 2025 8:07 AM
|
Machine learning has supercharged the field of computational protein design.
|
|
Scooped by
Gerd Moe-Behrens
January 16, 2025 8:06 AM
|
Digital art techniques can now devise custom, working biomolecules on demand.
|
|
Scooped by
Gerd Moe-Behrens
November 1, 2024 8:50 PM
|
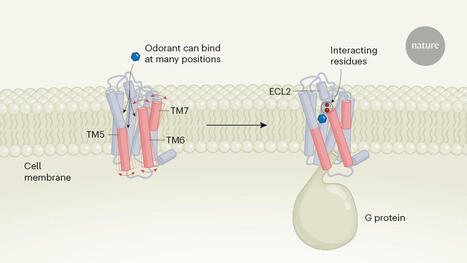
The structures of engineered odorant receptors.
|
|
Scooped by
Gerd Moe-Behrens
September 8, 2024 2:15 PM
|
Multi-layered computational gene networks by engineered tristate logics
|
|
Scooped by
Gerd Moe-Behrens
July 12, 2024 7:52 AM
|
Microbiome research is now demonstrating a growing number of bacterial strains and genes that affect our health1. Although CRISPR-derived tools have shown great success in editing disease-driving genes in human cells2, we currently lack the tools to achieve comparable success for bacterial targets in situ. Here we engineer a phage-derived particle to deliver a base editor and modify Escherichia coli colonizing the mouse gut. Editing of a β-lactamase gene in a model E. coli strain resulted in a median editing efficiency of 93% of the target bacterial population with a single dose. Edited bacteria were stably maintained in the mouse gut for at least 42 days following treatment. This was achieved using a non-replicative DNA vector, preventing maintenance and dissemination of the payload. We then leveraged this approach to edit several genes of therapeutic relevance in E. coli and Klebsiella pneumoniae strains in vitro and demonstrate in situ editing of a gene involved in the production of curli in a pathogenic E. coli strain. Our work demonstrates the feasibility of modifying bacteria directly in the gut, offering a new avenue to investigate the function of bacterial genes and opening the door to the design of new microbiome-targeted therapies. Edited bacteria were stably maintained in mouse gut for at least 42 days following the delivery of a base editor using an engineered phage-derived particle to modify Escherichia coli colonizing the gut.
|
|
Scooped by
Gerd Moe-Behrens
July 12, 2024 7:50 AM
|
Plasmids are indispensable in life sciences research and therapeutics development. Currently, most labs custom-build their plasmids. As yet, no systematic data on the quality of lab-made plasmids exist. Here, we report a broad survey of plasmids from hundreds of academic and industrial labs worldwide. We show that nearly half of them contained design and/or sequence errors. For transfer plasmids used in making AAV vectors, which are widely used in gene therapy, about 40% carried mutations in ITR regions due to their inherent instability, which is influenced by flanking GC content. We also list genes difficult to clone into plasmid or package into virus due to their toxicity. Our finding raises serious concerns over the trustworthiness of lab-made plasmids, which parallels the underappreciated mycoplasma contamination and misidentified mammalian cell lines reported previously, and highlights the need for community-wide standards to uphold the quality of this ubiquitous reagent in research and medicine. https://www.biorxiv.org/content/10.1101/2024.06.17.596931v1
|
|
Scooped by
Gerd Moe-Behrens
June 27, 2024 3:29 PM
|
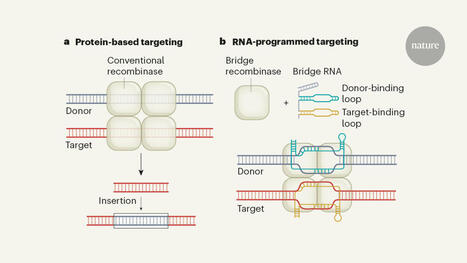
Recombinase enzymes that recognize DNA sequences using a ‘bridge’ RNA.
|