Your new post is loading...
Your new post is loading...

|
Scooped by
Dr. Stefan Gruenwald
November 26, 2023 9:47 AM
|
A versatile, scalable search engine for genomic and sequencing project metadata across the eukaryotic tree of life. Projects GoaT hosts dedicated pages for the Earth Biogenome Project (EBP) and EBP-affiliated projects. Each project page contains information about the project, including its GoaT search term and BioProject ID together with reports of highlighting the planning, progress and completion of genome assemblies. Earth Biogenome Project EBP Affiliated Project Networks
|
|
Scooped by
Dr. Stefan Gruenwald
March 14, 2023 12:01 PM
|
The Cancer Genome Atlas (TCGA), a landmark cancer genomics program, molecularly characterized over 20,000 primary cancer and matched normal samples spanning 33 cancer types. This joint effort between NCI and the National Human Genome Research Institute began in 2006, bringing together researchers from diverse disciplines and multiple institutions. Over the next dozen years, TCGA generated over 2.5 petabytes of genomic, epigenomic, transcriptomic, and proteomic data. The data, which has already led to improvements in our ability to diagnose, treat, and prevent cancer, will remain publicly available for anyone in the research community to use.
|
|
Scooped by
Dr. Stefan Gruenwald
July 13, 2022 10:49 AM
|
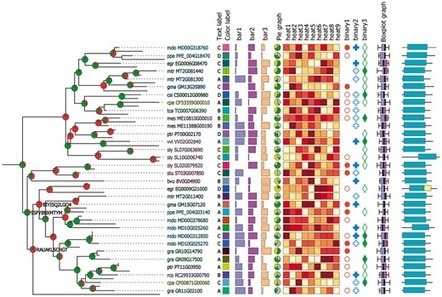
Phylogenetic trees showing the early diversification of fungi This study shows a Bayesian consensus tree inferred from a concatenated alignment of 299 proteins and 101 taxa under the CAT+GTR+Γ4 (CATGTR in the text) model. Node support is given by posterior probabilities).
Diversity of fungal phyla. FLTR, first row: Ascomycota, Cookeina colensoi (https://mushroomobserver.org); Basidiomycota, Amanita muscaria (https://mushroomobserver.org); Glomeromycota, Glomus sp. (https://comenius.susqu.edu). Second row: Mucoromycota, Pilobolus kleinii (https://fungi.myspecies.info); Mortierellomycota, Mortierella elongata (https://mycocosm.jgi.doe.gov, credit: Kerry L. O’Donnell); Zoopagomycota, Piptocephalis sp. (https://en.wikipedia.org). Third row: Olpidiomycota, Olpidium virulentus (https://ephytia.inra.fr); Blastocladiomycota, Allomyces sp. (https://mushroomobserver.org); Sanchytriomycota, Sanchytrium tribonematis (Galindo et al.10). Fourth row: Chytridiomycota, Synchytridium endobioticum (https://en.wikipedia.org); Neocallimastigomycota, Neocallimastix californiae (https://mycocosm.jgi.doe.gov, credit: John Henske); Monoblepharomycota, Gonapodya prolifera (https://mycocosm.jgi.doe.gov, credit: Jaclyn Dee). Fifth row: Aphelidiomycota, Paraphelidium tribonematis (Torruella et al.18); Microsporidia, Spraguea lophii (https://mycocosm.jgi.doe.gov, credit: Bryony Williams); Rozellomycota, Rozella allomycis (https://en.wikipedia.org).
|
|
Rescooped by
Dr. Stefan Gruenwald
from DNA and RNA research
December 13, 2017 2:49 PM
|
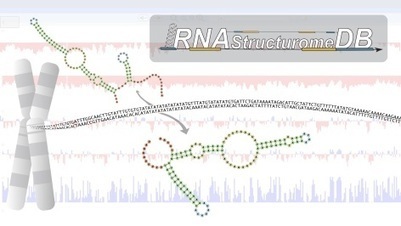
Iowa State University researchers have built an accessible online database that brings critical genomic data into sharp focus with the single click of a mouse. In an article published today in the journal Scientific Reports, a team of Iowa State University researchers presented a novel database that allows scientists to quickly access information on RNA structures encoded within the human genome. The database is freely accessible to anyone on the web, where it allows scientists to study the functions and structure of RNA with greater speed and ease than in the past. This will help facilitate basic research into human biology and could pave the way for new medical treatments that target RNA, said Walter Moss, an assistant professor in the Roy J. Carver Department of Biochemistry, Biophysics and Molecular Biology.
RNAs, short for ribonucleic acids, are molecular chains that are transcribed, or copied, from the genome. RNAs can be information carriers, acting as intermediates between DNA and protein, or as independently functioning molecules that mediate many critical regulatory processes. Unlike DNA, RNA can “fold back” on itself to form structures that rival proteins in their complexity. This folding plays key roles in RNA function, so gaining structural information on RNA can provide important insights into our understanding of how organisms work, Moss said.
“RNA is fundamental to understanding biology, and advances in sequencing technology have led to an explosion in the list of potentially functional RNAs,” he said.
For example, while only about 2 percent of the 3.5 billion base pairs of human DNA encode proteins, more than 70 percent is transcribed into RNA. Understanding the functions of this ocean of uncharted RNA poses a key scientific challenge, and the new database provides crucial tools for charting it, Moss said.
Moss, along with graduate student Ryan Andrews and Research IT Director Levi Baber, fragmented the entire human genome into more than 154 million overlapping windows and calculated a range of metrics valuable in understanding how RNA folds.
“Anyone around the world can now retrieve with one click what once took hours of calculations to determine,” Andrews said.
RNA therapeutics, a new approach in medicine in which RNA is targeted with drugs rather than proteins, show great promise, and the structural database detailed in the publication provides a huge repository of potential therapeutic leads, Moss said.
Via Integrated DNA Technologies
|
|
Rescooped by
Dr. Stefan Gruenwald
from The science toolbox
October 11, 2017 12:50 PM
|
Comparative and evolutionary studies utilize phylogenetic trees to analyze and visualize biological data.
Via Niklaus Grunwald
|
|
Scooped by
Dr. Stefan Gruenwald
September 11, 2017 1:49 PM
|
The Bioinformatics Links Directory features curated links to molecular resources, tools and databases. The links listed in this directory are selected on the basis of recommendations from bioinformatics experts in the field. Starting in 2003, all links contained in the NAR Webserver issue are included.
|
|
Scooped by
Dr. Stefan Gruenwald
September 8, 2017 12:32 PM
|
Oomycetes Transcriptomics Database is an integrated transcriptome and EST data resource for oomycete pathogens. The database currently stores processed ABI SOLiD transcript sequences from Phytophthora sojae mycelial and plant infection libraries as well as Illumina transcript sequences from five Hyaloperonospora arabidopsidis libraries. In addition to those resources, it has also a complete set of Sanger EST sequences from P. sojae, P. infestans and H. arabidopsidis grown under various conditions. A new web-based transcriptome browser was created for visualization of assembled transcripts, their mapping to the reference genome, expression profiling and depth of read coverage for a particular location on the genome. The transcriptome browser merges EST derived contigs with NGS derived assembled transcripts on the fly and displays the consensus. OTD possesses strong query features and the database interacts with VBI Microbial Database as well as the Phytophthora Transcriptomics Database. The legacy EST data of P.sojae comes from 10 libraries e.g; sHA, sHB, sMA, sML, sMY, sMC, sZG, sZO, sZS, iMY with a total of 33350 raw sequences. Additionally, there are 99,320 EST sequences fromP.infestans from NCBI. For P.infestans sequences cleaning information is not available. These sequences are clustered and assembled and data analysis was performed by us. We have separated Soybean ESTs from P.sojae libraries using insillico methods. The soybean libraries are named as gHA and gHB based on their origin from sHA or sHB.
|
|
Scooped by
Dr. Stefan Gruenwald
April 15, 2016 4:38 PM
|
Expression compendia in M3D are versioned to indicate the underlying mysql schema of the database and to denote the particular set of expression data (e.g. E_coli_v4_Build_6 uses mysql schema version 4 and is the sixth compendium built for E. coli). Builds are maintained in perpetuity, allowing researchers to specify the exact dataset used for a particular analysis. nt
|
|
Scooped by
Dr. Stefan Gruenwald
April 15, 2016 12:48 PM
|
|
|
Scooped by
Dr. Stefan Gruenwald
April 15, 2016 12:42 PM
|
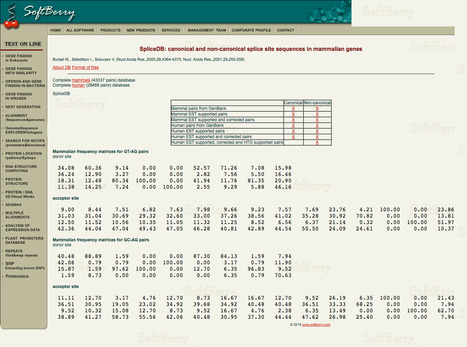
A set of 43337 splice junction pairs was extracted from mammalian GenBank annotated genes. 22489 of them are supported by EST sequences. 98.71% of those contain canonical dinucleotides GT and AG for donor and acceptor sites, respectively. 0.56% hold non-canonical GC-AG splice site pairs. The reminder 0.73% occurs in a lot of small groups (with maximum size of 0.05%). Studying these groups we observe that many of them contain splicing dinucleotides shifted from the annotated splice junction by one position. After close examination of such cases we present a new classification consisting of only 8 observed types of splice site pairs (out of 256 a priori possible combinations).
|
|
Scooped by
Dr. Stefan Gruenwald
April 15, 2016 12:39 PM
|
L1Resources Database This web portal is dedicated to putatively active LINE-1 elements. LINE-1 (Long Interspersed Nucleotide Element 1, L1) are the only autonomous retrotransposons in the group of non-LTR retro-transposons. They comprise about 18%-20% of mammalian genomes.
|
|
Scooped by
Dr. Stefan Gruenwald
April 15, 2016 12:34 PM
|
Select parts and Make Embeddable Model of Your Own.
|
|
Scooped by
Dr. Stefan Gruenwald
April 6, 2016 4:23 PM
|
In the post genome era, proteomic technology has rapidly developed to be a powerful platform for the research of human physiology and identifying potential novel biomarkers for prognosis, diagnosis and therapeusis . And in recent years it is shown that body fluids has become one of the important targets for proteomic research. Analysis of the protein composition in body fluids could help to better understand human physiology and disease proteomics. More and more proteomics research have produced body fluid related data and a database is needed to collect and analyze these data. Our body fluid protein database, Sys-BodyFluid, contains 11 body fluid proteomes, including plasma/serum, urine, cerebrospinal fluid, saliva, bronchoalveolar lavage fluid, synovial fluid, nipple aspirate fluid, tear fluid, seminal fluid, human milk, and amniotic fluid. Over 10,000 proteins are included in the Sys-BodyFluid. These body fluid proteome data come from 50 peer-review publications of different laboratories all over the world. Protein annotation are provided including protein description, Gene ontology, Domain information, Protein sequence and involved pathway. User can access the proteome data by protein name, protein accession number, sequence similarity. In addition, user could perform query cross different body fluids to get more comprehensive understanding. The difference and similarity between these 11 body fluids are also analyzed. Thus , the Sys-BodyFluid database could serve as a reference database for body fluid research and disease proteomics. Click here to edit the content
|
|
|
Scooped by
Dr. Stefan Gruenwald
October 12, 2023 9:07 PM
|
Alphabetical listing is here The 2023 Nucleic Acids Research Database Issue contains 178 papers ranging across biology and related fields. There are 90 papers reporting on new databases and 82 updates from resources previously published in the Issue. Six more papers are updates from databases most recently published elsewhere. Major nucleic acid databases reporting updates include Genbank, ENA, ChIPBase, JASPAR, mirDIP and the Issue's first Breakthrough Article, NACDDB for Circular Dichroism data. Updates from BMRB and RCSB cover experimental protein structural data while AlphaFold 2 computational structure predictions feature widely. STRING and REBASE are stand-out updates in the signalling and enzymes section. Immunology-related databases include CEDAR, the second Breakthrough Article, for cancer epitopes and receptors alongside returning IPD-IMGT/HLA and the new PGG.MHC. Genomics-related resources include Ensembl, GWAS Central and UCSC Genome Browser. Major returning databases for drugs and their targets include Open Targets, DrugCentral, CTD and Pubchem. The EMPIAR image archive appears in the Issue for the first time. The entire database Issue is freely available online on the Nucleic Acids Research website (https://academic.oup.com/nar). The NAR online Molecular Biology Database Collection has been updated, revisiting 463 entries, adding 92 new resources and eliminating 96 discontinued URLs so bringing the current total to 1764 databases.
|
|
Scooped by
Dr. Stefan Gruenwald
March 2, 2023 10:29 AM
|
The GlycoNAVI database is a tool for carbohydrate researchers. These databases are linked to various glycan related databases. This database link is linked not only to glycans but to the life-science database, and researchers can obtain various information. GlycoNAVI is a chemical abbreviation database which contains abbreviations of functional groups and major chemical compounds.
|
|
Rescooped by
Dr. Stefan Gruenwald
from Plants and Microbes
November 13, 2018 12:59 PM
|
The Plant Genome Editing Database currently provides information about plants that have been generated using the CRISPR/Cas9 technology in order to study economically important traits. Users begin by either choosing the species they are interested in and browsing the list of genes that carry mutations or searching the database using specific gene identifiers (e.g., Solyc05g053230). Information provided includes the transformation experiment, the name of the transformed plant variety, the DNA construct used including the guide RNA sequence and primers used to characterize resulting mutations, and details about the mutant plant line including the altered sequence, whether it is heterozygous or homozygous, and any phenotypes that have been observed. Users are encouraged to make information available about their own CRISPR-generated plant lines and details are provided about how data can be submitted for inclusion in the database.
Via Kamoun Lab @ TSL
|
|
Scooped by
Dr. Stefan Gruenwald
November 10, 2017 2:14 PM
|
NAR database issue online (for 2017)
|
|
Scooped by
Dr. Stefan Gruenwald
September 11, 2017 2:16 PM
|
Software packages for next gen sequence analysis Bioinformatics Integrated solutions
* CLCbio Genomics Workbench - de novo and reference assembly of Sanger, Roche FLX, Illumina, Helicos, and SOLiD data. Commercial next-gen-seq software that extends the CLCbio Main Workbench software. Includes SNP detection, CHiP-seq, browser and other features. Commercial. Windows, Mac OS X and Linux.
* Galaxy - Galaxy = interactive and reproducible genomics. A job webportal.
* Genomatix - Integrated Solutions for Next Generation Sequencing data analysis.
* JMP Genomics - Next gen visualization and statistics tool from SAS. They are working with NCGR to refine this tool and produce others.
* NextGENe - de novo and reference assembly of Illumina, SOLiD and Roche FLX data. Uses a novel Condensation Assembly Tool approach where reads are joined via "anchors" into mini-contigs before assembly. Includes SNP detection, CHiP-seq, browser and other features. Commercial. Win or MacOS.
* SeqMan Genome Analyser - Software for Next Generation sequence assembly of Illumina, Roche FLX and Sanger data integrating with Lasergene Sequence Analysis software for additional analysis and visualization capabilities. Can use a hybrid templated/de novo approach. Commercial. Win or Mac OS X.
* SHORE - SHORE, for Short Read, is a mapping and analysis pipeline for short DNA sequences produced on a Illumina Genome Analyzer. A suite created by the 1001 Genomes project. Source for POSIX.
* SlimSearch - Fledgling commercial product.
Align/Assemble to a reference
* BFAST - Blat-like Fast Accurate Search Tool. Written by Nils Homer, Stanley F. Nelson and Barry Merriman at UCLA.
* Bowtie - Ultrafast, memory-efficient short read aligner. It aligns short DNA sequences (reads) to the human genome at a rate of 25 million reads per hour on a typical workstation with 2 gigabytes of memory. Uses a Burrows-Wheeler-Transformed (BWT) index.Link to discussion thread here. Written by Ben Langmead and Cole Trapnell. Linux, Windows, and Mac OS X.
* BWA - Heng Lee's BWT Alignment program - a progression from Maq. BWA is a fast light-weighted tool that aligns short sequences to a sequence database, such as the human reference genome. By default, BWA finds an alignment within edit distance 2 to the query sequence. C++ source.
* ELAND - Efficient Large-Scale Alignment of Nucleotide Databases. Whole genome alignments to a reference genome. Written by Illumina author Anthony J. Cox for the Solexa 1G machine.
* Exonerate - Various forms of pairwise alignment (including Smith-Waterman-Gotoh) of DNA/protein against a reference. Authors are Guy St C Slater and Ewan Birney from EMBL. C for POSIX.
* GenomeMapper - GenomeMapper is a short read mapping tool designed for accurate read alignments. It quickly aligns millions of reads either with ungapped or gapped alignments. A tool created by the 1001 Genomes project. Source for POSIX.
* GMAP - GMAP (Genomic Mapping and Alignment Program) for mRNA and EST Sequences. Developed by Thomas Wu and Colin Watanabe at Genentec. C/Perl for Unix.
* gnumap - The Genomic Next-generation Universal MAPper (gnumap) is a program designed to accurately map sequence data obtained from next-generation sequencing machines (specifically that of Solexa/Illumina) back to a genome of any size. It seeks to align reads from nonunique repeats using statistics. From authors at Brigham Young University. C source/Unix.
* MAQ - Mapping and Assembly with Qualities (renamed from MAPASS2). Particularly designed for Illumina with preliminary functions to handle ABI SOLiD data. Written by Heng Li from the Sanger Centre. Features extensive supporting tools for DIP/SNP detection, etc. C++ source
* MOSAIK - MOSAIK produces gapped alignments using the Smith-Waterman algorithm. Features a number of support tools. Support for Roche FLX, Illumina, SOLiD, and Helicos. Written by Michael Strömberg at Boston College. Win/Linux/MacOSX
* MrFAST and MrsFAST - mrFAST & mrsFAST are designed to map short reads generated with the Illumina platform to reference genome assemblies; in a fast and memory-efficient manner. Robust to INDELs and MrsFAST has a bisulphite mode. Authors are from the University of Washington. C as source.
* MUMmer - MUMmer is a modular system for the rapid whole genome alignment of finished or draft sequence. Released as a package providing an efficient suffix tree library, seed-and-extend alignment, SNP detection, repeat detection, and visualization tools. Version 3.0 was developed by Stefan Kurtz, Adam Phillippy, Arthur L Delcher, Michael Smoot, Martin Shumway, Corina Antonescu and Steven L Salzberg - most of whom are at The Institute for Genomic Research in Maryland, USA. POSIX OS required.
* Novocraft - Tools for reference alignment of paired-end and single-end Illumina reads. Uses a Needleman-Wunsch algorithm. Can support Bis-Seq. Commercial. Available free for evaluation, educational use and for use on open not-for-profit projects. Requires Linux or Mac OS X.
* PASS - It supports Illumina, SOLiD and Roche-FLX data formats and allows the user to modulate very finely the sensitivity of the alignments. Spaced seed intial filter, then NW dynamic algorithm to a SW(like) local alignment. Authors are from CRIBI in Italy. Win/Linux.
* RMAP - Assembles 20 - 64 bp Illumina reads to a FASTA reference genome. By Andrew D. Smith and Zhenyu Xuan at CSHL. (published in BMC Bioinformatics). POSIX OS required.
* SeqMap - Supports up to 5 or more bp mismatches/INDELs. Highly tunable. Written by Hui Jiang from the Wong lab at Stanford. Builds available for most OS's.
* SHRiMP - Assembles to a reference sequence. Developed with Applied Biosystem's colourspace genomic representation in mind. Authors are Michael Brudno and Stephen Rumble at the University of Toronto. POSIX.
* Slider- An application for the Illumina Sequence Analyzer output that uses the probability files instead of the sequence files as an input for alignment to a reference sequence or a set of reference sequences. Authors are from BCGSC. Paper is here.
* SOAP - SOAP (Short Oligonucleotide Alignment Program). A program for efficient gapped and ungapped alignment of short oligonucleotides onto reference sequences. The updated version uses a BWT. Can call SNPs and INDELs. Author is Ruiqiang Li at the Beijing Genomics Institute. C++, POSIX.
* SSAHA - SSAHA (Sequence Search and Alignment by Hashing Algorithm) is a tool for rapidly finding near exact matches in DNA or protein databases using a hash table. Developed at the Sanger Centre by Zemin Ning, Anthony Cox and James Mullikin. C++ for Linux/Alpha.
* SOCS - Aligns SOLiD data. SOCS is built on an iterative variation of the Rabin-Karp string search algorithm, which uses hashing to reduce the set of possible matches, drastically increasing search speed. Authors are Ondov B, Varadarajan A, Passalacqua KD and Bergman NH.
* SWIFT - The SWIFT suit is a software collection for fast index-based sequence comparison. It contains: SWIFT — fast local alignment search, guaranteeing to find epsilon-matches between two sequences. SWIFT BALSAM — a very fast program to find semiglobal non-gapped alignments based on k-mer seeds. Authors are Kim Rasmussen (SWIFT) and Wolfgang Gerlach (SWIFT BALSAM)
* SXOligoSearch - SXOligoSearch is a commercial platform offered by the Malaysian based Synamatix. Will align Illumina reads against a range of Refseq RNA or NCBI genome builds for a number of organisms. Web Portal. OS independent.
* Vmatch - A versatile software tool for efficiently solving large scale sequence matching tasks. Vmatch subsumes the software tool REPuter, but is much more general, with a very flexible user interface, and improved space and time requirements. Essentially a large string matching toolbox. POSIX.
* Zoom - ZOOM (Zillions Of Oligos Mapped) is designed to map millions of short reads, emerged by next-generation sequencing technology, back to the reference genomes, and carry out post-analysis. ZOOM is developed to be highly accurate, flexible, and user-friendly with speed being a critical priority. Commercial. Supports Illumina and SOLiD data.
De novo Align/Assemble
* ABySS - Assembly By Short Sequences. ABySS is a de novo sequence assembler that is designed for very short reads. The single-processor version is useful for assembling genomes up to 40-50 Mbases in size. The parallel version is implemented using MPI and is capable of assembling larger genomes. By Simpson JT and others at the Canada's Michael Smith Genome Sciences Centre. C++ as source.
* ALLPATHS - ALLPATHS: De novo assembly of whole-genome shotgun microreads. ALLPATHS is a whole genome shotgun assembler that can generate high quality assemblies from short reads. Assemblies are presented in a graph form that retains ambiguities, such as those arising from polymorphism, thereby providing information that has been absent from previous genome assemblies. Broad Institute.
* Edena - Edena (Exact DE Novo Assembler) is an assembler dedicated to process the millions of very short reads produced by the Illumina Genome Analyzer. Edena is based on the traditional overlap layout paradigm. By D. Hernandez, P. François, L. Farinelli, M. Osteras, and J. Schrenzel. Linux/Win.
* EULER-SR - Short read de novo assembly. By Mark J. Chaisson and Pavel A. Pevzner from UCSD (published in Genome Research). Uses a de Bruijn graph approach.
* MIRA2 - MIRA (Mimicking Intelligent Read Assembly) is able to perform true hybrid de-novo assemblies using reads gathered through 454 sequencing technology (GS20 or GS FLX). Compatible with 454, Solexa and Sanger data. Linux OS required.
* SEQAN - A Consistency-based Consensus Algorithm for De Novo and Reference-guided Sequence Assembly of Short Reads. By Tobias Rausch and others. C++, Linux/Win.
* SHARCGS - De novo assembly of short reads. Authors are Dohm JC, Lottaz C, Borodina T and Himmelbauer H. from the Max-Planck-Institute for Molecular Genetics.
* SSAKE - The Short Sequence Assembly by K-mer search and 3' read Extension (SSAKE) is a genomics application for aggressively assembling millions of short nucleotide sequences by progressively searching for perfect 3'-most k-mers using a DNA prefix tree. Authors are René Warren, Granger Sutton, Steven Jones and Robert Holt from the Canada's Michael Smith Genome Sciences Centre. Perl/Linux.
* SOAPdenovo - Part of the SOAP suite. See above.
* VCAKE - De novo assembly of short reads with robust error correction. An improvement on early versions of SSAKE.
* Velvet - Velvet is a de novo genomic assembler specially designed for short read sequencing technologies, such as Solexa or 454. Need about 20-25X coverage and paired reads. Developed by Daniel Zerbino and Ewan Birney at the European Bioinformatics Institute (EMBL-EBI).
SNP/Indel Discovery
* ssahaSNP - ssahaSNP is a polymorphism detection tool. It detects homozygous SNPs and indels by aligning shotgun reads to the finished genome sequence. Highly repetitive elements are filtered out by ignoring those kmer words with high occurrence numbers. More tuned for ABI Sanger reads. Developers are Adam Spargo and Zemin Ning from the Sanger Centre. Compaq Alpha, Linux-64, Linux-32, Solaris and Mac
* PolyBayesShort - A re-incarnation of the PolyBayes SNP discovery tool developed by Gabor Marth at Washington University. This version is specifically optimized for the analysis of large numbers (millions) of high-throughput next-generation sequencer reads, aligned to whole chromosomes of model organism or mammalian genomes. Developers at Boston College. Linux-64 and Linux-32.
* PyroBayes - PyroBayes is a novel base caller for pyrosequences from the 454 Life Sciences sequencing machines. It was designed to assign more accurate base quality estimates to the 454 pyrosequences. Developers at Boston College.
Genome Annotation/Genome Browser/Alignment Viewer/Assembly Database
* EagleView - An information-rich genome assembler viewer. EagleView can display a dozen different types of information including base quality and flowgram signal. Developers at Boston College.
* LookSeq - LookSeq is a web-based application for alignment visualization, browsing and analysis of genome sequence data. LookSeq supports multiple sequencing technologies, alignment sources, and viewing modes; low or high-depth read pileups; and easy visualization of putative single nucleotide and structural variation. From the Sanger Centre.
* MapView - MapView: visualization of short reads alignment on desktop computer. From the Evolutionary Genomics Lab at Sun-Yat Sen University, China. Linux.
* SAM - Sequence Assembly Manager. Whole Genome Assembly (WGA) Management and Visualization Tool. It provides a generic platform for manipulating, analyzing and viewing WGA data, regardless of input type. Developers are Rene Warren, Yaron Butterfield, Asim Siddiqui and Steven Jones at Canada's Michael Smith Genome Sciences Centre. MySQL backend and Perl-CGI web-based frontend/Linux.
* STADEN - Includes GAP4. GAP5 once completed will handle next-gen sequencing data. A partially implemented test version is available here
* XMatchView - A visual tool for analyzing cross_match alignments. Developed by Rene Warren and Steven Jones at Canada's Michael Smith Genome Sciences Centre. Python/Win or Linux.
Counting e.g. CHiP-Seq, Bis-Seq, CNV-Seq
* BS-Seq - The source code and data for the "Shotgun Bisulphite Sequencing of the Arabidopsis Genome Reveals DNA Methylation Patterning" Nature paper by Cokus et al. (Steve Jacobsen's lab at UCLA). POSIX.
* CHiPSeq - Program used by Johnson et al. (2007) in their Science publication
* CNV-Seq - CNV-seq, a new method to detect copy number variation using high-throughput sequencing. Chao Xie and Martti T Tammi at the National University of Singapore. Perl/R.
* FindPeaks - perform analysis of ChIP-Seq experiments. It uses a naive algorithm for identifying regions of high coverage, which represent Chromatin Immunoprecipitation enrichment of sequence fragments, indicating the location of a bound protein of interest. Original algorithm by Matthew Bainbridge, in collaboration with Gordon Robertson. Current code and implementation by Anthony Fejes. Authors are from the Canada's Michael Smith Genome Sciences Centre. JAVA/OS independent. Latest versions available as part of the Vancouver Short Read Analysis Package
* MACS - Model-based Analysis for ChIP-Seq. MACS empirically models the length of the sequenced ChIP fragments, which tends to be shorter than sonication or library construction size estimates, and uses it to improve the spatial resolution of predicted binding sites. MACS also uses a dynamic Poisson distribution to effectively capture local biases in the genome sequence, allowing for more sensitive and robust prediction. Written by Yong Zhang and Tao Liu from Xiaole Shirley Liu's Lab.
* PeakSeq - PeakSeq: Systematic Scoring of ChIP-Seq Experiments Relative to Controls. a two-pass approach for scoring ChIP-Seq data relative to controls. The first pass identifies putative binding sites and compensates for variation in the mappability of sequences across the genome. The second pass filters out sites that are not significantly enriched compared to the normalized input DNA and computes a precise enrichment and significance. By Rozowsky J et al. C/Perl.
* QuEST - Quantitative Enrichment of Sequence Tags. Sidow and Myers Labs at Stanford. From the 2008 publication Genome-wide analysis of transcription factor binding sites based on ChIP-Seq data. (C++)
* SISSRs - Site Identification from Short Sequence Reads. BED file input. Raja Jothi @ NIH. Perl.
**See also this thread for ChIP-Seq, until I get time to update this list.
Alternate Base Calling
* Rolexa - R-based framework for base calling of Solexa data. Project publication
* Alta-cyclic - "a novel Illumina Genome-Analyzer (Solexa) base caller"
Transcriptomics
* ERANGE - Mapping and Quantifying Mammalian Transcriptomes by RNA-Seq. Supports Bowtie, BLAT and ELAND. From the Wold lab.
* G-Mo.R-Se - G-Mo.R-Se is a method aimed at using RNA-Seq short reads to build de novo gene models. First, candidate exons are built directly from the positions of the reads mapped on the genome (without any ab initio assembly of the reads), and all the possible splice junctions between those exons are tested against unmapped reads. From CNS in France.
* MapNext - MapNext: A software tool for spliced and unspliced alignments and SNP detection of short sequence reads. From the Evolutionary Genomics Lab at Sun-Yat Sen University, China.
* QPalma - Optimal Spliced Alignments of Short Sequence Reads. Authors are Fabio De Bona, Stephan Ossowski, Korbinian Schneeberger, and Gunnar Rätsch. A paper is available.
* RSAT - RSAT: RNA-Seq Analysis Tools. RNASAT is developed and maintained by Hui Jiang at Stanford University.
* TopHat - TopHat is a fast splice junction mapper for RNA-Seq reads. It aligns RNA-Seq reads to mammalian-sized genomes using the ultra high-throughput short read aligner Bowtie, and then analyzes the mapping results to identify splice junctions between exons. TopHat is a collaborative effort between the University of Maryland and the University of California, Berkeley
|
|
Scooped by
Dr. Stefan Gruenwald
September 8, 2017 1:05 PM
|
Translation initiation sites that precede the main coding sequence (CDS) of a transcript give rise to upstream open reading frames (uORFs). The presence of uORFs affects initiation rates at the CDS by interfering with unrestrained progression of ribosomes across the 5´-transcript leader sequence.
|
|
Scooped by
Dr. Stefan Gruenwald
September 8, 2017 12:16 PM
|
FungiDB provides functional genomics of fungi.
|
|
Scooped by
Dr. Stefan Gruenwald
April 15, 2016 3:51 PM
|
Gene Annotations Various classes of gene annotations (e.g. functions, pathways, diseases association, tissue expression, etc) from Lynx Knowledge base integrating information from over 40 public databasest. Enrichment Analysis Identification of gene categories over-represented in the user-submitted gene list based on Pathways, Gene Ontology, Diseases, Tissue expression and other features associations. Genes Prioritization Networks based Gene Prioritization using PINTA algorithms: Heat Kernal Ranking, Simple Random Walk, PageRank with Priors, HITS with Priors and K-Step Markov. Using STRING-9 underlying network.
|
|
Scooped by
Dr. Stefan Gruenwald
April 15, 2016 12:44 PM
|
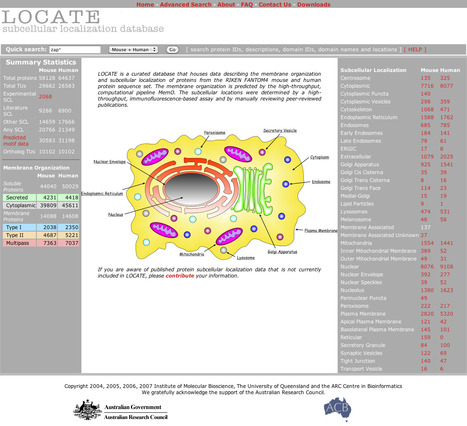
LOCATE is a curated database that houses data describing the membrane organization and subcellular localization of proteins from the RIKEN FANTOM4 mouse and human protein sequence set. The membrane organization is predicted by the high-throughput, computational pipeline MemO. The subcellular locations were determined by a high-throughput, immunofluorescence-based assay and by manually reviewing peer-reviewed publications.
|
|
Scooped by
Dr. Stefan Gruenwald
April 15, 2016 12:41 PM
|
PlantProm DB was initially developed as an annotated, non-redundant collection of proximal promoter sequences for RNA polymerase II with experimentally determined transcription start site(s), TSS, from various plant species. The first release of DB, 2002.01, developed by the Department of Computer Science at Royal Holloway, University of London, in collaboration with Softberry Inc. (USA), is available at http://mendel.cs.rhul.ac.uk/mendel.php . It contained 305 entries from monocot, dicot and other plants.
The new release of PlantProm DB contains 578 unrelated entries including 151, 396 and 31 promoters with experimentally verified TSS from monocot, dicot and other plants, respectively.In comparison with promoter sets, where TSSs, identified by applying full-length cDNA/5;-5'ESTs mapping, CAGE and SAGE approaches, remain to be confirmed by direct experimental evidence, this DB and The Eukaryotic Promoter Database (134 unrelated plant promoters; see:http://www.epd.isb-sib.ch/ ) present the published promoter sequences with TSS(s) determined by direct experimental approaches and therefore serve as the most accurate sources for development of computational promoter prediction tools (for example, see: TSSP-TCM, TSSP, FPROM, CONPRO). For collecting experimentally verified plant gene promoters the following criteria was followed.
|
|
Scooped by
Dr. Stefan Gruenwald
April 15, 2016 12:35 PM
|
Herbal Ingredients' Targets Database HIT is a comprehensive and fully curated database to complement available resources on protein targets for FDA-approved drugs as well as the promising precursors. It currently contains about 1,301 known protein targets(221 proteins are described as direct targets) derived from more than 3,250 literatures, which covers about 586 active compounds from more than 1,300 reputable Chinese herbs. The molecular target information involves those proteins being directly/indirectly activated or inhibited, protein binders, and enzymes whose substrates or products are those compounds. Detailed interaction values such as IC50 and Kd/Ki are collected if possible. Those up or down regulated genes are also included under the treatment of individual ingredients.
|
|
Scooped by
Dr. Stefan Gruenwald
April 6, 2016 4:25 PM
|
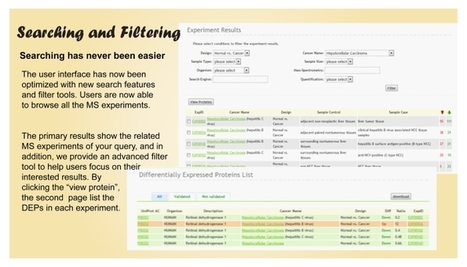
dbDEPC is updated to version 2.0 and contains more information about differentially expressed proteins (DEPs) in human cancers. Currently dbDEPC contains 4029 DEPs, 20 cancers and 331 MS experiments. More advanced search function makes your query easier. Modified profile and new network function provide you a systematic view of DEPs in cancers.
|