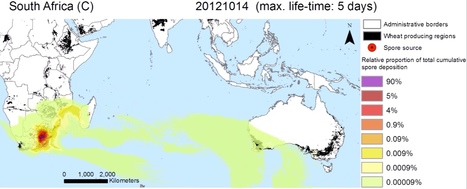
The Australian wheat stem rust (Puccinia graminis f. sp. tritici) population was shaped by the introduction of four exotic incursions into the country. It was previously hypothesized that at least two of these (races 326-1,2,3,5,6 and 194-1,2,3,5,6 first detected in 1969) had an African origin and moved across the Indian Ocean to Australia on high-altitude winds. We provide strong supportive evidence for this hypothesis by combining genetic analyses and complex atmospheric dispersion modeling. Genetic analysis of 29 Australian and South African P. graminis f. sp. tritici races using microsatellite markers confirmed the close genetic relationship between the South African and Australian populations, thereby confirming previously described phenotypic similarities. Lagrangian particle dispersion model simulations using finely resolved meteorological data showed that long distance dispersal events between southern Africa and Australia are indeed possible, albeit rare. Simulated urediniospore transmission events were most frequent from central South Africa (viable spore transmission on approximately 7% of all simulated release days) compared with other potential source regions in southern Africa. The study acts as a warning of possible future P. graminis f. sp. tritici dispersal events from southern Africa to Australia, which could include members of the Ug99 race group, emphasizing the need for continued surveillance on both continents.
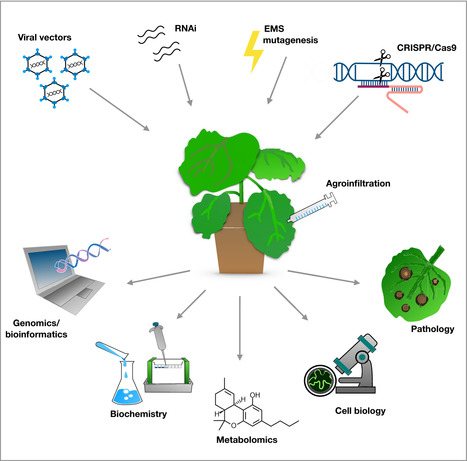
Nicotiana benthamiana, a solanaceous species native to Australia, is one of the most commonly used model plant organisms. It was initially used by the virology community because of its hyper‐susceptibility to plant viruses. Later, this feature was exploited with the development of viral vectors that can express foreign genes and the establishment of virus‐induced gene silencing (VIGS), which enabled knocking down endogenous plant genes. Benth (or Benthi), as it is colloquially known, was further popularized by the development of agroinfiltration, a method that enabled transient protein expression in plants. Agroinfiltration has been extensively used in cell biology, biochemistry, protein–protein interaction analyses and other in planta studies (Goodin et al., 2008). However, genetic analyses of N. benthamiana remained limited due to its allotetraploid nature and incomplete sequence of the 3.1 Gb genome. In this issue of New Phytologist, Schultink et al. (pp. 1001–1009) used a forward genetic screen in N. benthamiana to determine that NbZAR1, an orthologue of the Arabidopsis thaliana NLR (nucleotide‐binding domain and leucine‐rich repeat domain‐containing) ZAR1, is responsible for the perception of XopJ4 from the tomato bacterial pathogen Xanthomonas perforans. Although forward genetic screens using gene silencing have been performed in N. benthamiana, Schultink et al. are among the first to use a chemical mutagenesis‐based screen to dissect plant biological processes. Together with CRISPR genome editing and improved genomics resources, this study ushers in a new era of forward and reverse genetic analyses for this much‐cherished model plant system (Fig. 1).
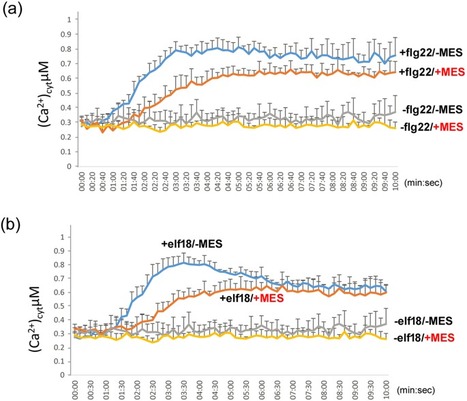
Agrobacterium-mediated transient expression is a powerful analysis platform for diverse plant gene functional studies, but the mechanisms regulating the expression or transformation levels are poorly studied. Previously, we developed a highly efficient and robust Agrobacterium-mediated transient expression system, named AGROBEST, for Arabidopsis seedlings. In this study, we found that AGROBEST could promote the growth of agrobacteria as well as inhibit the host immunity response. When the factor of agrobacterial growth is minimized, maintaining pH at 5.5 with MES buffer was the key to achieving optimal transient expression efficiency. The expression of plant immunity marker genes, FRK1 and NHL10, was suppressed in the pH-buffered medium as compared with non-buffered conditions in Col-0 and an efr-1 mutant lacking the immunity receptor EFR recognizing EF-Tu, a potent pathogen- or microbe-associated molecular pattern (PAMP or MAMP) of A. tumefaciens. Notably, such immune suppression could also occur in Arabidopsis seedlings without Agrobacterium infection. Furthermore, the PAMP-triggered influx of calcium ions was compromised in the pH-buffered medium. We propose that the enhanced transient expression efficiency by stable pH was due to inhibiting calcium ion uptake and subsequently led to suppressing immunity against Agrobacterium.
Downy mildew, once just a nuisance, has evolved into a devastating adversary to the pickle, as it now quickly adapts to fungicides and pickle hybrids, and can lay waste to crops in a matter of days.
The pickle is in peril. Each summer since the mid 2000s, Florida winds carry downy mildew to cucumber fields north. By summer's end, the disease reaches Michigan, leaving a trail of withered leaves and thwarted pickling plans.
With failed harvests, fewer growers are taking a chance on cucumbers. According to USDA records, pickling cucumber acreage declined nearly 25 percent between 2004 and 2015. Globally, downy mildew threatens fields as far flung as India, Israel, Mexico and China.
"This is the number one threat to the pickle industry," says vegetable pathologist Lina Quesada-Ocampo of North Carolina State University. The growers, she says, lose money on failed crops and pricey fungicides. "It is a really bad double whammy."
Fortunately for pickle lovers, vegetable breeder Michael Mazourek of Cornell University is close to releasing varieties that resist downy mildew. "It's been one of our proudest David and Goliath stories," he says. But his success hinges on funding at a time when public support of agricultural research is declining.
The story of saving the pickle, then, is not just about preserving the deli sandwich's sidekick. It's a story of how much we value our food supply. And who we think should pay to protect it.
The ability to induce a defense response after pathogen attack is a critical feature of the immune system of any organism. Nucleotide-binding leucine-rich repeat receptors (NLRs) are key players in this process and perceive the occurrence of nonself-activities or foreign molecules. In plants, coevolution with a variety of pests and pathogens has resulted in repertoires of several hundred diverse NLRs in single individuals and many more in populations as a whole. However, the mechanism by which defense signaling is triggered by these NLRs in plants is poorly understood. Here, we show that upon pathogen perception, NLRs use their N-terminal domains to transactivate other receptors. Their N-terminal domains homo- and heterodimerize, suggesting that plant NLRs oligomerize upon activation, similar to the vertebrate NLRs; however, consistent with their large number in plants, the complexes are highly heterometric. Also, in contrast to metazoan NLRs, the N-terminus, rather than their centrally located nucleotide-binding (NB) domain, can mediate initial partner selection. The highly redundant network of NLR interactions in plants is proposed to provide resilience to perturbation by pathogens.
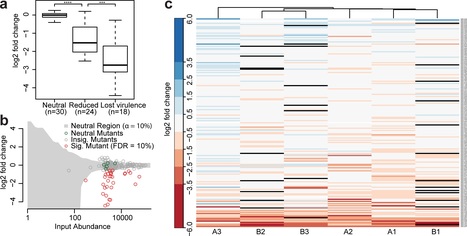
Large-scale insertional mutagenesis screens can be powerful genome-wide tools if they are streamlined with efficient downstream analysis, which is a serious bottleneck in complex biological systems. A major impediment to the success of next-generation sequencing (NGS)-based screens for virulence factors is that the genetic material of pathogens is often underrepresented within the eukaryotic host, making detection extremely challenging. We therefore established insertion Pool-Sequencing (iPool-Seq) on maize infected with the biotrophic fungus U. maydis. iPool-Seq features tagmentation, unique molecular barcodes, and affinity purification of pathogen insertion mutant DNA from in vivo-infected tissues. In a proof of concept using iPool-Seq, we identified 28 virulence factors, including 23 that were previously uncharacterized, from an initial pool of 195 candidate effector mutants. Because of its sensitivity and quantitative nature, iPool-Seq can be applied to any insertional mutagenesis library and is especially suitable for genetically complex setups like pooled infections of eukaryotic hosts.
"I have always been fascinated by how beautiful and diverse plants are." Stella Cesari is the winner of the 2017 New Phytologist Tansley Medal for excellence in plant science.
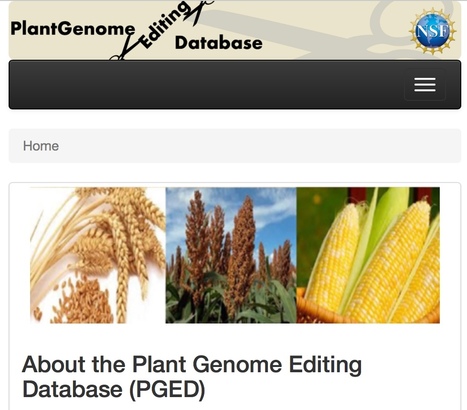
The Plant Genome Editing Database currently provides information about plants that have been generated using the CRISPR/Cas9 technology in order to study economically important traits. Users begin by either choosing the species they are interested in and browsing the list of genes that carry mutations or searching the database using specific gene identifiers (e.g., Solyc05g053230). Information provided includes the transformation experiment, the name of the transformed plant variety, the DNA construct used including the guide RNA sequence and primers used to characterize resulting mutations, and details about the mutant plant line including the altered sequence, whether it is heterozygous or homozygous, and any phenotypes that have been observed. Users are encouraged to make information available about their own CRISPR-generated plant lines and details are provided about how data can be submitted for inclusion in the database.
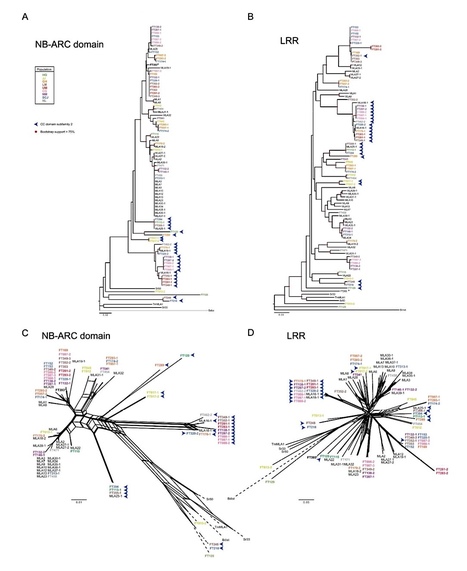
The barley disease resistance (R) gene locus Mildew Locus A (Mla) provides isolate-specific resistance against the powdery mildew fungus Blumeria graminis hordei (Bgh) and has been introgressed into modern cultivars from diverse germplasms, including the wild relative Hordeum spontaneum. Known Mla disease resistance specificities to Bgh appear to encode allelic variants of the R Gene Homolog 1 (RGH1) family of nucleotide-binding domain and leucine-rich repeat (NLR) proteins. We here sequenced and assembled the transcriptomes of 50 H. spontaneum accessions representing nine populations distributed throughout the Fertile Crescent. The assembled Mla transcripts exhibited rich sequence diversity, linked neither to geographic origin nor population structure and could be grouped into two similar-sized subfamilies based on two major N-terminal coiled-coil signaling domains that are both capable of eliciting cell death. The presence of positively selected sites, located mainly in the C-terminal leucine-rich repeats of both MLA subfamilies, together with the fact that both coiled-coil signaling domains mediate cell death, implies that the two subfamilies are actively maintained in the population. Unexpectedly, known MLA receptor variants that confer Bghresistance belong exclusively to one subfamily. Thus, signaling domain divergence, potentially as adaptation to distinct pathogen populations, is an evolutionary signature of functional diversification of an immune receptor.
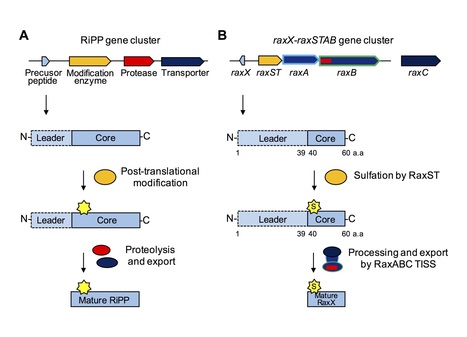
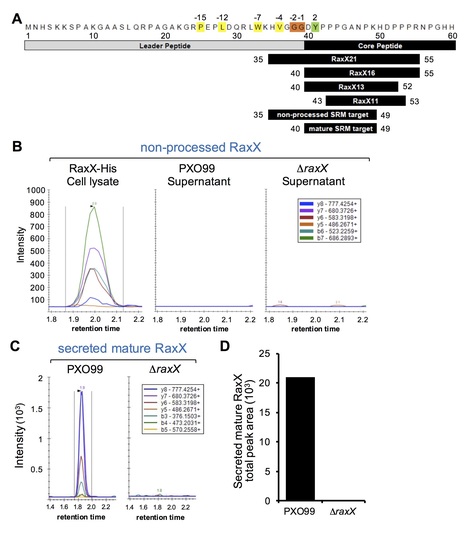
The rice immune receptor XA21 is activated by the sulfated microbial peptide RaxX (required for activation of XA21-mediated immunity X) produced by Xanthomonas oryzae pv. oryzae (Xoo). Mutational studies and targeted proteomics revealed that RaxX is processed and secreted by the protease/transporter RaxB, whose function can be partially fulfilled by a noncognate peptidase-containing transporter B (PctB). RaxX is cleaved at a Gly-Gly motif, yielding a mature peptide that retains the necessary elements for RaxX function as an immunogen and host peptide hormone mimic. These results indicate that RaxX is a founding member of a previously unclassified and understudied group of tyrosine sulfated RiPPs (ribosomally synthesized, post-translationally modified peptides). We further demonstrate that sulfated RaxX directly binds XA21 with high affinity. This work reveals a complete, previously uncharacterized biological process: bacterial RiPP biosynthesis, secretion, binding to a eukaryotic receptor and triggering of a robust host immune response.
Author summary Plants are constantly exposed to a multitude of potential pathogens but remain immune to most of these due to a multilayered immune system. Pathogens have specialized by adapting to certain host plants and their defense barriers. Most of our understanding of plant-pathogen interactions stems from these highly specialized interactions, because they are characterized by qualitative interactions (resistant or susceptible). It has generally been assumed that the genetic and molecular basis of resistance to non-adapted pathogens is fundamentally different, as either no variation exists in a species (complete immunity) or variation encompasses only early pathogen invasion (colonization), but not full susceptibility. We have studied the interaction between the agronomically important fungal stripe rust pathogen (Puccinia striiformis) of wheat and barley with the wild grass species Brachypodium distachyon. Rust infections consist of two stages: colonization of plant tissues followed by a reproductive phase. We identified natural variation for the degree of P. striiformis colonization in different B. distachyon accessions and dissected the genetic architecture controlling resistance at this infection stage. QTLs conferring resistance possessed several characteristics similar to adapted host systems, indicating that resistance to adapted and non-adapted pathogens are not intrinsically different.
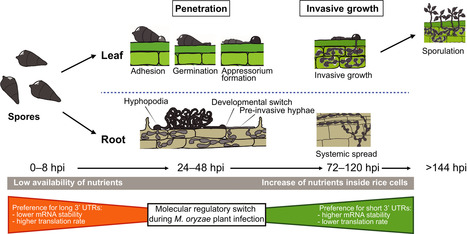
Generation of mRNA isoforms by alternative polyadenylation (APA) and their involvement in regulation of fungal cellular processes, including virulence, remains elusive. Here, we investigated genome‐wide polyadenylation site (PAS) selection in the rice blast fungus to understand how APA regulates pathogenicity.
More than half of Magnaporthe oryzae transcripts undergo APA and show novel motifs in their PAS region. Transcripts with shorter 3′UTRs are more stable and abundant in polysomal fractions, suggesting they are being translated more efficiently. Importantly, rice colonization increases the use of distal PASs of pathogenicity genes, especially those participating in signalling pathways like 14‐3‐3B, whose long 3′UTR is required for infection.
Cleavage factor I (CFI) Rbp35 regulates expression and distal PAS selection of virulence and signalling‐associated genes, tRNAs and transposable elements, pointing its potential to drive genomic rearrangements and pathogen evolution. We propose a noncanonical PAS selection mechanism for Rbp35 that recognizes UGUAH, unlike humans, without CFI25.
Our results showed that APA controls turnover and translation of transcripts involved in fungal growth and environmental adaptation. Furthermore, these data provide useful information for enhancing genome annotations and for cross‐species comparisons of PASs and PAS usage within the fungal kingdom and the tree of life.
Interactions: What guided your decision to dedicate the next stage of your research career to MPMI?
Detlef Weigel: My path to MPMI was rather circuitous. Genetics is my first love, and genetic phenomena of any kind appeal to me. Almost 15 years ago, Janne Lempe and Kirsten Bomblies in my lab discovered a syndrome of Arabidopsis hybrid weakness that we at first interpreted as a developmental abnormality. We quickly learned that this syndrome was not specific to Arabidopsis spp. and that it was already well known from many wild and cultivated plants, for which it is called “hybrid necrosis.” Anybody in the MPMI field knows that necrosis is often a hallmark of pathogen infection. Nevertheless, we were apparently the first ones to recognize that inappropriate immune reactions in the absence of pathogens were most likely the defining characteristics of this phenomenon, rather than developmental defects.
For us, one of the attractions of studying hybrid necrosis was that we thought it would teach us about speciation, but after many thousands of crosses and having cloned quite a few of the causal genes, we realized that hybrid necrosis has much more to do with how the plant balances the demands on its immune system. With too little immunity, the plant will succumb too quickly to infection, but with too much immunity, the plant will damage itself. Hybrid necrosis occurs when components of the immune system are mismatched, and these components begin to signal even if there is no pathogen trigger. Satisfyingly, the molecular observations in Arabidopsis spp. seemed to match similar observations in several other species. As a matter of fact, with hindsight we realized that the first case of hybrid necrosis that was molecularly understood predated our own work in Arabidopsis—namely, the study of the tomato Cf-2/Rcr3 system by Jonathan Jones.
In parallel with our efforts to clone the causal genes for hybrid necrosis in Arabidopsis spp., we could confirm through our whole-genome resequencing and sequencing studies that immune genes—particularly those of the NLR class but also of other smaller families—are the most diverse genes in the Arabidopsis genome. This, in turn, made us wonder what drives this diversity—hence, our current obsession with trying to understand the relationship between Arabidopsis and its natural pathogens in the real world.
The 14-3-3 phospho-binding proteins with scaffolding activity play central roles in the regulation of enzymes and signaling complexes in eukaryotes. In plants, 14-3-3 isoforms are required for disease resistance and key targets of pathogen effectors. Here, we examined the requirement of the tomato (Solanum lycopersicum) 14-3-3 isoform (TFT) protein family for Xv3 disease resistance in response to the bacterial pathogen Xanthomonas euvesicatoria. In addition, we determined whether TFT proteins interact with the repertoire of X. euvesicatoria type III secretion effector proteins, including AvrXv3, the elicitor of Xv3 resistance. We show that multiple TFT contribute to Xv3resistance. We also show that one or more TFT proteins physically interact with multiple effectors (AvrXv3, XopE1, XopE2, XopN, XopO, XopQ, and XopAU). Genetic analyses indicate that none of the identified effectors interfere with AvrXv3-elicited resistance into Xv3 tomato leaves; however, XopE1, XopE2, and XopO are required to suppress symptom development in susceptible tomato leaves. Phospho-peptide mapping revealed that XopE2 is phosphorylated at multiple residues in planta and residues T66, T131, and S334 are required for maximal binding to TFT10. Together, our data support the hypothesis that multiple TFT proteins are involved in immune signaling during X. euvesicatoria infection.
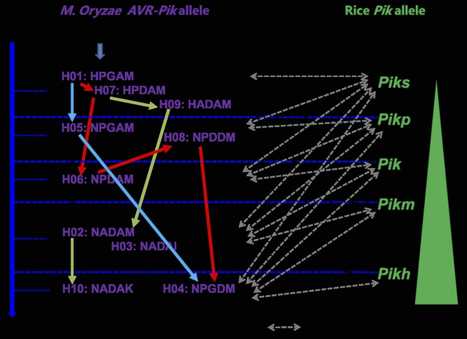
Rice blast disease is one of the most destructive fungal diseases of rice world-wide. The avirulence (AVR) genes of Magnaporthe oryzae are recognized by the cognate resistance (R) genes of rice, and trigger race specific resistance. Here, we studied the possible evolutionary pathways in the evolution of AVR-Pik alleles by analyzing the DNA sequence variation and assayed for their avirulence function to the cognate Pik alleles resistance genes under field conditions in China. Results of PCR products showed that 278 isolates of M. oryzae carry AVR-Pik alleles among genomic DNA of 366 isolates of M. oryzae collected from Yunnan Province, China. Among of them, 66.7-90.3% of M. oryzae carry AVR-Pik alleles from six regions of Yunnan. Moreover, 10 AVR-Pik haplotypes encoding five novel AVR-Pik variants were identified among 201 isolates. The AVR-Pik alleles stepwise evolved to virulence from avirulent forms via base substitution. These findings demonstrate that AVR-Pik alleles are under positive selection and mutations are responsible for defeating race-specific resistance Pik alleles in nature.
The rice immune receptor XA21 is activated by the sulfated microbial peptide RaxX (required for activation of XA21-mediated immunity X) produced by Xanthomonas oryzae pv. oryzae (Xoo). Mutational studies and targeted proteomics revealed that RaxX is processed and secreted by the protease/transporter RaxB, whose function can be partially fulfilled by a noncognate peptidase-containing transporter B (PctB). RaxX is cleaved at a Gly-Gly motif, yielding a mature peptide that retains the necessary elements for RaxX function as an immunogen and host peptide hormone mimic. These results indicate that RaxX is a founding member of a previously unclassified and understudied group of tyrosine sulfated RiPPs (ribosomally synthesized, post-translationally modified peptides). We further demonstrate that sulfated RaxX directly binds XA21 with high affinity. This work reveals a complete, previously uncharacterized biological process: bacterial RiPP biosynthesis, secretion, binding to a eukaryotic receptor and triggering of a robust host immune response.
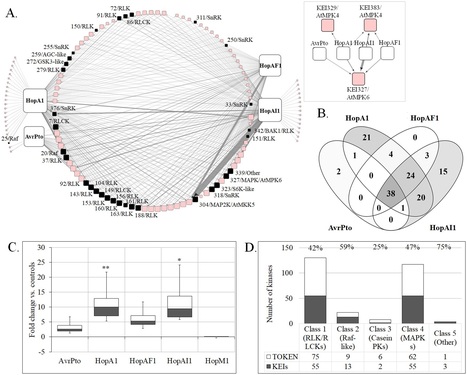
Author summary Some bacterial pathogens secrete virulence factors called effectors, which influence host tissues during infection. The impact of such bacterial effectors on the transmission of immune signals in plants remains poorly understood. In this study, we adapted an integrative network approach to discover interactions between bacterial effectors and a class of host signal-mediating enzymes called protein kinases. We also characterized the functions of the targets of these kinases in order to understand how bacterial effectors might disrupt the flow of information in signaling pathways within plant cells. We show that plants activate larger signaling networks when inoculated with pathogens that produce effectors. We also find that plant signaling networks are specific to individual effectors and that the networks include kinases with both positive and negative effects on plant resistance to pathogens. We propose that the topology of immune signaling networks is determined by the plant’s ability to activate compensatory pathways in response to the effectors’ network-disruptive actions. Conversely, pathogens may increase their virulence both by disrupting host signaling at the membrane-located end of the signaling network and by recruiting cytosolic kinases. This work provides a framework for the study of plant–pathogen communication and could be used to prioritize targets for improving resistance in crops.
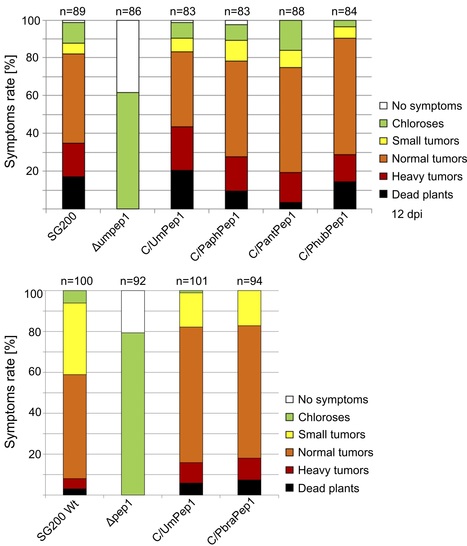
The basidiomycete smut fungi are predominantly plant parasitic, causing severe losses in some crops. Most species feature a saprotrophic haploid yeast stage, and several smut fungi are only known from this stage, with some isolated from habitats without suitable hosts, e.g. from Antarctica. Thus, these species are generally believed to be apathogenic, but recent findings that some of these might have a plant pathogenic sexual counterpart, casts doubts on the validity of this hypothesis. Here, four Pseudozyma genomes were re-annotated and compared to published smut pathogens and the well-characterised effector gene Pep1 from these species was checked for its ability to complement a Pep1 deletion strain of Ustilago maydis. It was found that 113 high-confidence putative effector proteins were conserved among smut and Pseudozyma genomes. Among these were several validated effector proteins, including Pep1. By genetic complementation we show that Pep1 homologs from the supposedly apathogenic yeasts restore virulence in Pep1-deficient mutants Ustilago maydis. Thus, it is concluded that Pseudozyma species have retained a suite of effectors. This hints at the possibility that Pseudozyma species have kept an unknown plant pathogenic stage for sexual recombination or that these effectors have positive effects when colonising plant surfaces.
While making the roadmap ”wish-list” for the future, the last item, “food”, set the plant biologist in Dr. Naweed Naqvi thinking about what could be the major challenges faced by food crops or plants in general, in the future. In this talk, he speaks about how we can engineer precision plant-immunity by studying the defence pathways of rice, a simple, but powerful crop. Naweed Naqvi obtained his PhD from the Maharaja Sayajirao University of Baroda (India) and the International Rice Research Institute (IRRI, Philippines) in 1995. He worked as a Rockefeller Foundation Fellow at IRRI until 1997, when he moved to the Institute of Molecular Agrobiology, Singapore for a postdoctoral stint in eukaryotic cell division. He subsequently started his independent research group at IMA focusing primarily on Fungal Pathogenesis. He joined the Temasek Life Sciences Laboratory as a Senior Scientist in 2002, and serves as an Adjunct Professor at the National University of Singapore This talk was given at a TEDx event using the TED conference format but independently organized by a local community. Click here to edit the contentClick here to edit the content
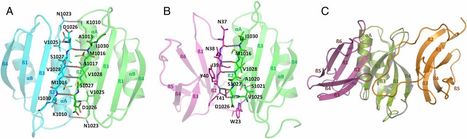
The structurally conserved but sequence-unrelated MAX (Magnaporthe oryzae avirulence and ToxB-like) effectors AVR1-CO39 and AVR-PikD from the blast fungus M. oryzae are recognized by the rice nucleotide-binding domain and leucine-rich repeat proteins (NLRs) RGA5 and Pikp-1, respectively. This involves, in both cases, direct interaction of the effector with a heavy metal-associated (HMA) integrated domain (ID) in the NLR. Here, we solved the crystal structures of a C-terminal fragment of RGA5 carrying the HMA ID (RGA5_S), alone, and in complex with AVR1-CO39 and compared it to the structure of the Pikp1HMA/AVR-PikD complex. In both complexes, HMA ID/MAX effector interactions involve antiparallel alignment of β-sheets from each partner. However, effector-binding occurs at different surfaces in Pikp1HMA and RGA5HMA, indicating that these interactions evolved independently by convergence of these two MAX effectors to the same type of plant target proteins. Interestingly, the effector-binding surface in RGA5HMA overlaps with the surface that mediates RGA5HMA self-interaction. Mutations in the HMA-binding interface of AVR1-CO39 perturb RGA5HMA-binding, in vitro and in vivo, and affect the recognition of M. oryzae in a rice cultivar containing Pi-CO39. Our study provides detailed insight into the mechanisms of effector recognition by NLRs, which has substantial implications for future engineering of NLRs to expand their recognition specificities. In addition, we propose, as a hypothesis for the understanding of effector diversity, that in the structurally conserved MAX effectors the molecular mechanism of host target protein-binding is conserved rather than the host target proteins themselves.
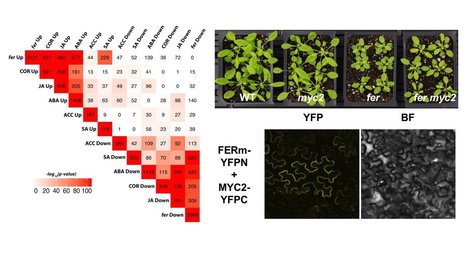
Bacterial pathogens use effectors and phytotoxins to facilitate infection of host plants. Coronatine (COR) is one of the phytotoxins produced in bacterial pathogens, such as Pseudomonas syringae pv. tomato DC3000 (pst DC3000). COR structurally and functionally mimics the active form of the plant hormone jasmonic acid (JA), JA-isoleucine (JA-Ile), and can hijack the host JA-signaling pathway to achieve host disease susceptibility [ 1 ]. COR utilizes the transcription factor MYC2, a master regulator of JA signaling, to activate NAC transcription factors, which functions to inhibit accumulation of salicylic acid (SA) and thus compromise host immunity [ 2 ]. It has been demonstrated that SA can antagonize JA signaling through NONEXPRESSOR of PATHOGENESIS-RELATED GENE1 (NPR1) [ 3 ] and downstream transcription factors TGAs [ 4 ] and WRKYs [ 5 , 6 ]. However, the detailed mechanism by which host plants counteract COR-mediated susceptibility is largely unknown. Here, we show that the receptor kinase FERONIA (FER) functions to inhibit JA and COR signaling by phosphorylating and destabilizing MYC2, thereby positively regulating immunity. Conversely, the peptide ligand RALF23 acts through FER to stabilize MYC2 and elevate JA signaling, negatively contributing to plant immunity. Our results establish the RALF23-FER-MYC2 signaling module and provide a previously unknown mechanism by which host plants utilize FER signaling to counteract COR-mediated host disease susceptibility.
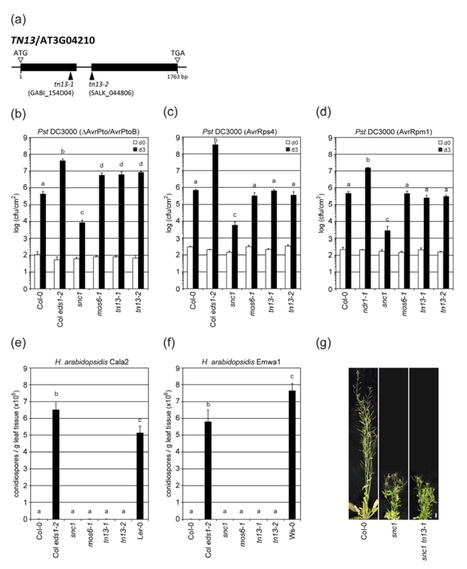
Importin‐α proteins mediate the translocation of nuclear localization signal (NLS)‐containing proteins from the cytoplasm into the nucleus through nuclear pore complexes (NPCs). Genetically, Arabidopsis IMPORTIN‐α3/MOS6 (MODIFIER OF SNC1, 6) is required for basal plant immunity and constitutive disease resistance activated in the autoimmune mutant snc1 (suppressor of npr1‐1, constitutive 1), suggesting that MOS6 plays a role in the nuclear import of proteins involved in plant defense signaling. Here, we sought to identify and characterize defense‐regulatory cargo proteins and interaction partners of MOS6. We conducted both in silico database analyses and affinity purification of functional epitope‐tagged MOS6 from pathogen‐challenged stable transgenic plants coupled with mass spectrometry. We show that among the 13 candidate MOS6 interactors we selected for further functional characterization, the TIR‐NBS‐type protein TN13 is required for resistance against Pseudomonas syringae pv. tomato (Pst) DC3000 lacking the type‐III effector proteins AvrPto and AvrPtoB. When expressed transiently in N. benthamianaleaves, TN13 co‐immunoprecipitates with MOS6, but not with its closest homolog IMPORTIN‐α6, and localizes to the endoplasmic reticulum (ER), consistent with a predicted N‐terminal transmembrane domain in TN13. Our work uncovered the truncated NLR protein TN13 as a component of plant innate immunity that selectively binds to MOS6/IMPORTIN‐α3 in planta. We speculate that the release of TN13 from the ER membrane in response to pathogen stimulus, and its subsequent nuclear translocation, is important for plant defense signal transduction.Click here to edit the content
To get content containing either thought or leadership enter:
To get content containing both thought and leadership enter:
To get content containing the expression thought leadership enter:
You can enter several keywords and you can refine them whenever you want. Our suggestion engine uses more signals but entering a few keywords here will rapidly give you great content to curate.

 Your new post is loading...
Your new post is loading...