Your new post is loading...
Your new post is loading...
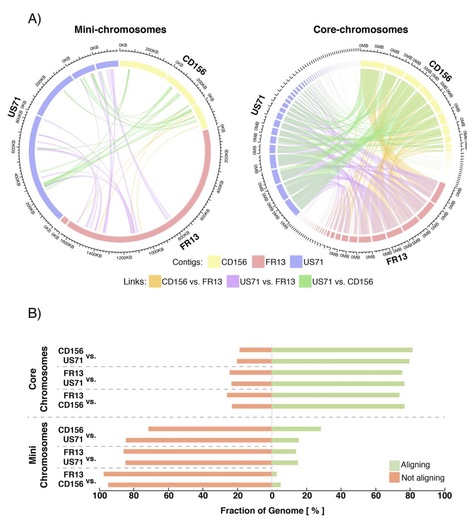
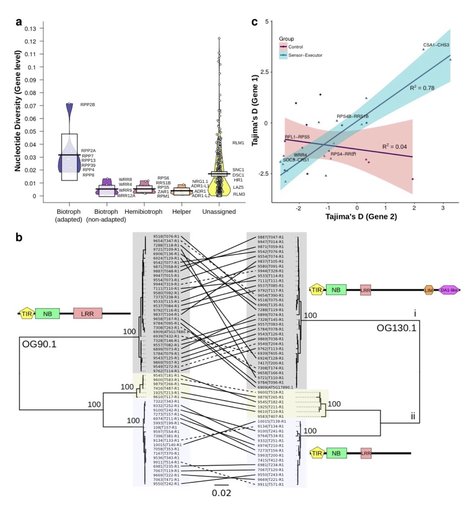
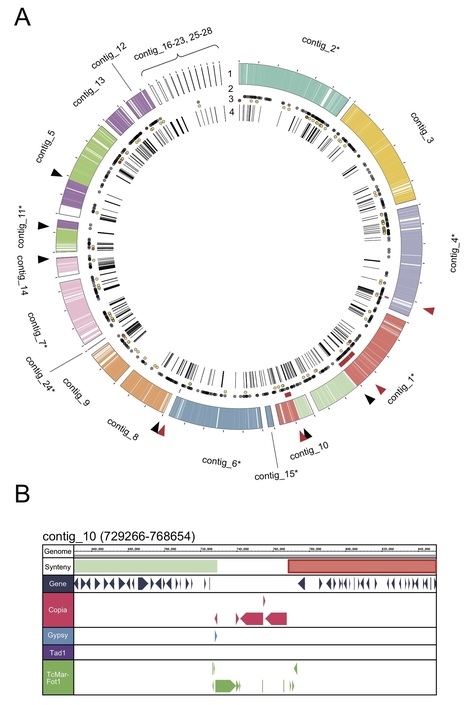
Supernumerary mini-chromosomes–a unique type of genomic structural variation–have been implicated in the emergence of virulence traits in plant pathogenic fungi. However, the mechanisms that facilitate the emergence and maintenance of mini-chromosomes across fungi remain poorly understood. In the blast fungus Magnaporthe oryzae, mini-chromosomes have been first described in the early 1990s but, until very recently, have been overlooked in genomic studies. Here we investigated structural variation in four isolates of the blast fungus M. oryzae from different grass hosts and analyzed the sequences of mini-chromosomes in the rice, foxtail millet and goosegrass isolates. The mini-chromosomes of these isolates turned out to be highly diverse with distinct sequence composition. They are enriched in repetitive elements and have lower gene density than core-chromosomes. We identified several virulence-related genes in the mini-chromosome of the rice isolate, including the polyketide synthase Ace1 and the effector gene AVR-Pik. Macrosynteny analyses around these loci revealed structural rearrangements, including inter-chromosomal translocations between core- and mini-chromosomes. Our findings provide evidence that mini-chromosomes independently emerge from structural rearrangements of core-chromosomes and might contribute to adaptive evolution of the blast fungus.
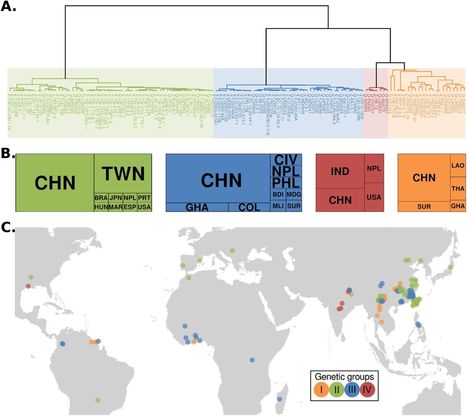
Background Understanding the mechanisms and timescales of plant pathogen outbreaks requires a detailed genome-scale analysis of their population history. The fungus Magnaporthe (Syn. Pyricularia) oryzae —the causal agent of blast disease of cereals— is among the most destructive plant pathogens to world agriculture and a major threat to the production of rice, wheat and other cereals. Although M. oryzae is a multihost pathogen that infects more than 50 species of cereals and grasses, all rice-infecting isolates belong to a single genetically defined lineage. Here, we combined multiple genomics datasets to reconstruct the genetic history of the rice-infecting lineage of M. oryzae based on 131 isolates from 21 countries. Results The global population of the rice blast fungus consists of a diverse set of individuals and three well-defined genetic groups. Multiple population genetic tests revealed that the rice-infecting lineage of the blast fungus probably originated from a recombining diverse group in South East Asia followed by three independent clonal expansions that took place over the last ∼200 years. Patterns of allele sharing identified a subpopulation from the recombining diverse group that introgressed with one of the clonal lineages before its global expansion. Remarkably, the four genetic lineages of the rice blast fungus vary in the number and patterns of presence/absence of candidate effector genes. In particular, clonal lineages carry a reduced repertoire of effector genes compared with the diverse group, and specific combinations of effector presence/absence define each of the pandemic clonal lineages. Conclusions Our analyses reconstruct the genetic history of the rice-infecting lineage of M. oryzae revealing three clonal lineages associated with rice blast pandemics. Each of these lineages displays a specific pattern of presence/absence of effector genes that may have shaped their adaptation to the rice host and their evolutionary history.
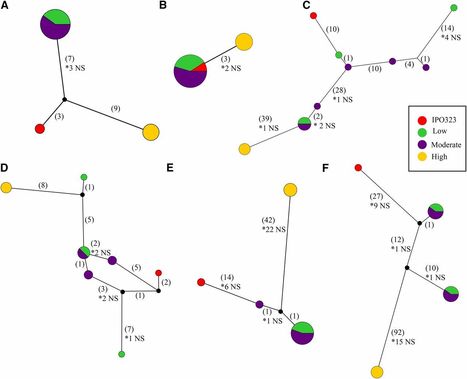
Zymoseptoria tritici is a host-specific, necrotrophic pathogen of wheat. Infection by Z. tritici is characterized by its extended latent period, which typically lasts 2 wks, and is followed by extensive host cell death, and rapid proliferation of fungal biomass. This work characterizes the level of genomic variation in 13 isolates, for which we have measured virulence on 11 wheat cultivars with differential resistance genes. Between the reference isolate, IPO323, and the 13 Australian isolates we identified over 800,000 single nucleotide polymorphisms, of which ∼10% had an effect on the coding regions of the genome. Furthermore, we identified over 1700 probable presence/absence polymorphisms in genes across the Australian isolates using de novo assembly. Finally, we developed a gene tree sorting method that quickly identifies groups of isolates within a single gene alignment whose sequence haplotypes correspond with virulence scores on a single wheat cultivar. Using this method, we have identified < 100 candidate effector genes whose gene sequence correlates with virulence toward a wheat cultivar carrying a major resistance gene.
Disease is both among the most important selection pressures in nature and among the main causes of yield loss in agriculture. In plants, resistance to disease is often conferred by Nucleotide-binding Leucine-rich Repeat (NLR) proteins. These proteins function as intracellular immune receptors that recognize pathogen proteins and their effects on the plant. Consistent with evolutionarily dynamic interactions between plants and pathogens, NLRs are known to be encoded by one of the most variable gene families in plants, but the true extent of intraspecific NLR diversity has been unclear. Here, we define the majority of the Arabidopsis thaliana species-wide 'NLRome'. From NLR sequence enrichment and long-read sequencing of 65 diverse A. thaliana accessions, we infer that the pan-NLRome saturates with approximately 40 accessions. Despite the high diversity of NLRs, half of the pan-NLRome is present in most accessions. We chart the architectural diversity of NLR proteins, identify novel architectures, and quantify the selective forces that act on specific NLRs, domains, and positions. Our study provides a blueprint for defining the pan-NLRome of plant species.
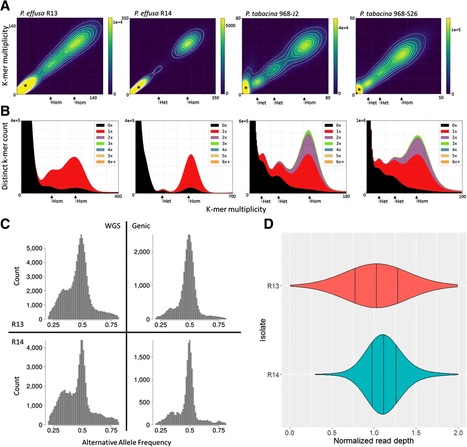
Background. Spinach downy mildew caused by the oomycete Peronospora effusa is a significant burden on the expanding spinach production industry, especially for organic farms where synthetic fungicides cannot be deployed to control the pathogen. P. effusa is highly variable and 15 new races have been recognized in the past 30 years. Results. We virulence phenotyped, sequenced, and assembled two isolates of P. effusa from the Salinas Valley, California, U.S.A. that were identified as race 13 and 14. These assemblies are high quality in comparison to assemblies of other downy mildews having low total scaffold count (784 & 880), high contig N50s (48 kb & 52 kb), high BUSCO completion and low BUSCO duplication scores and share many syntenic blocks with Phytophthora species. Comparative analysis of four downy mildew and three Phytophthora species revealed parallel absences of genes encoding conserved domains linked to transporters, pathogenesis, and carbohydrate activity in the biotrophic species. Downy mildews surveyed that have lost the ability to produce zoospores have a common loss of flagella/motor and calcium domain encoding genes. Our phylogenomic data support multiple origins of downy mildews from hemibiotrophic progenitors and suggest that common gene losses in these downy mildews may be of genes involved in the necrotrophic stages of Phytophthora spp. Conclusions. We present a high-quality draft genome of Peronospora effusa that will serve as a reference for Peronospora spp. We identified several Pfam domains as under-represented in the downy mildews consistent with the loss of zoosporegenesis and necrotrophy. Phylogenomics provides further support for a polyphyletic origin of downy mildews.
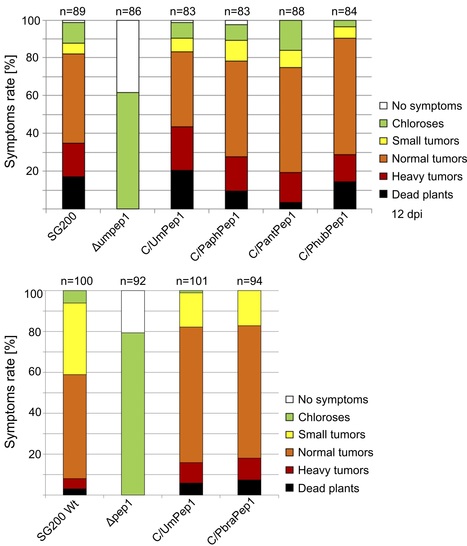
The basidiomycete smut fungi are predominantly plant parasitic, causing severe losses in some crops. Most species feature a saprotrophic haploid yeast stage, and several smut fungi are only known from this stage, with some isolated from habitats without suitable hosts, e.g. from Antarctica. Thus, these species are generally believed to be apathogenic, but recent findings that some of these might have a plant pathogenic sexual counterpart, casts doubts on the validity of this hypothesis. Here, four Pseudozyma genomes were re-annotated and compared to published smut pathogens and the well-characterised effector gene Pep1 from these species was checked for its ability to complement a Pep1 deletion strain of Ustilago maydis. It was found that 113 high-confidence putative effector proteins were conserved among smut and Pseudozyma genomes. Among these were several validated effector proteins, including Pep1. By genetic complementation we show that Pep1 homologs from the supposedly apathogenic yeasts restore virulence in Pep1-deficient mutants Ustilago maydis. Thus, it is concluded that Pseudozyma species have retained a suite of effectors. This hints at the possibility that Pseudozyma species have kept an unknown plant pathogenic stage for sexual recombination or that these effectors have positive effects when colonising plant surfaces.
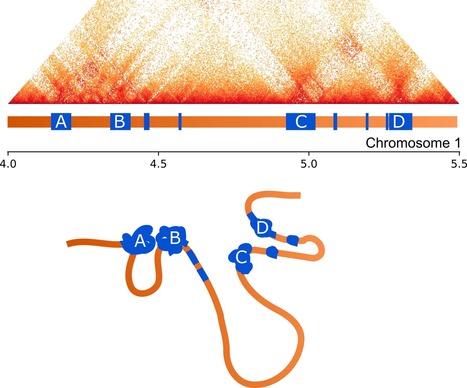
Author summary How a genome folds within a nucleus can contribute to the regulation of gene expression. There is now considerable interest in understanding how the three-dimensional structure of a genome is maintained and how this structure influences biological processes. However, most studies on these phenomena focus on a few model organisms, which limits our knowledge of how the nuclear organisation is maintained and the range of biological processes to which it contributes. In this paper, we examine the three-dimensional organisation of the genome in Epichloë festucae, a symbiotic fungus. Our results demonstrate that repetitive elements play a key role in shaping the nuclear organisation of E. festucae. These repetitive elements help to divide the genome into distinct regions that have similar gene expression profiles. Our results demonstrate a novel role for repeats in maintaining a three-dimensional organisation that contributes to regulating the symbiotic relationship formed by this species.
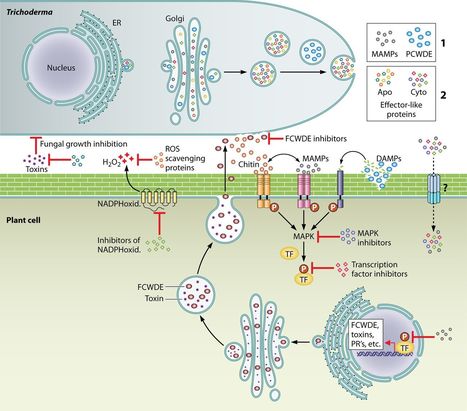
The genus Trichoderma contains fungi with high relevance for humans, with applications in enzyme production for plant cell wall degradation and use in biocontrol. Here, we provide a broad, comprehensive overview of the genomic content of these species for “hot topic” research aspects, including CAZymes, transport, transcription factors, and development, along with a detailed analysis and annotation of less-studied topics, such as signal transduction, genome integrity, chromatin, photobiology, or lipid, sulfur, and nitrogen metabolism in T. reesei, T. atroviride, and T. virens, and we open up new perspectives to those topics discussed previously. In total, we covered more than 2,000 of the predicted 9,000 to 11,000 genes of each Trichoderma species discussed, which is >20% of the respective gene content. Additionally, we considered available transcriptome data for the annotated genes. Highlights of our analyses include overall carbohydrate cleavage preferences due to the different genomic contents and regulation of the respective genes. We found light regulation of many sulfur metabolic genes. Additionally, a new Golgi 1,2-mannosidase likely involved in N-linked glycosylation was detected, as were indications for the ability of Trichoderma spp. to generate hybrid galactose-containing N-linked glycans. The genomic inventory of effector proteins revealed numerous compounds unique to Trichoderma, and these warrant further investigation. We found interesting expansions in the Trichoderma genus in several signaling pathways, such as G-protein-coupled receptors, RAS GTPases, and casein kinases. A particularly interesting feature absolutely unique to T. atroviride is the duplication of the alternative sulfur amino acid synthesis pathway.
Outbreaks caused by asexual lineages of fungal and oomycete pathogens are a continuing threat to crops, wild animals and natural ecosystems (Fisher MC, Henk DA, Briggs CJ, Brownstein JS, Madoff LC, McCraw SL, Gurr SJ, Nature 484:186–194, 2012; Kupferschmidt K, Science 337:636–638, 2012). However, the mechanisms underlying genome evolution and phenotypic plasticity in asexual eukaryotic microbes remain poorly understood (Seidl MF, Thomma BP, BioEssays 36:335–345, 2014). Ever since the 19th century Irish famine, the oomycete Phytophthora infestans has caused recurrent outbreaks on potato and tomato crops that have been primarily caused by the successive rise and migration of pandemic asexual lineages (Goodwin SB, Cohen BA, Fry WE, Proc Natl Acad Sci USA 91:11591–11595, 1994; Yoshida K, Burbano HA, Krause J, Thines M, Weigel D, Kamoun S, PLoS Pathog 10:e1004028, 2014; Yoshida K, Schuenemann VJ, Cano LM, Pais M, Mishra B, Sharma R, Lanz C, Martin FN, Kamoun S, Krause J, et al. eLife 2:e00731, 2013; Cooke DEL, Cano LM, Raffaele S, Bain RA, Cooke LR, Etherington GJ, Deahl KL, Farrer RA, Gilroy EM, Goss EM, et al. PLoS Pathog 8:e1002940, 2012). However, the dynamics of genome evolution within these clonal lineages have not been determined. The objective of this study was to use a comparative genomics and transcriptomics approach to determine the molecular mechanisms that underpin phenotypic variation within a clonal lineage of P. infestans.
Via The Sainsbury Lab
Following the molecular characterisation of functional disease resistance genes in recent years, methods to track and verify the integrity of multiple genes in varieties are needed for crop improvement through resistance stacking. Diagnostic resistance gene enrichment sequencing (dRenSeq) enables the high-confidence identification and complete sequence validation of known functional resistance genes in crops. As demonstrated for tetraploid potato varieties, the methodology is more robust and cost-effective in monitoring resistances than whole-genome sequencing and can be used to appraise (trans)gene integrity efficiently. All currently known NB-LRRs effective against viruses, nematodes and the late blight pathogen Phytophthora infestans can be tracked with dRenSeq in potato and hitherto unknown polymorphisms have been identified. The methodology provides a means to improve the speed and efficiency of future disease resistance breeding in crops by directing parental and progeny selection towards effective combinations of resistance genes.
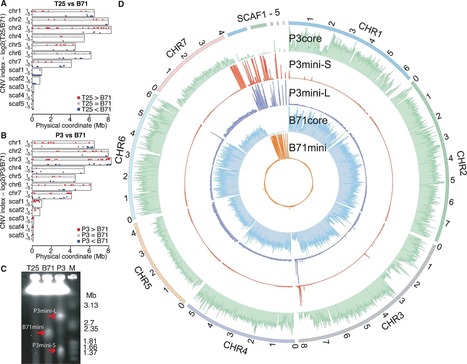
Newly emerged wheat blast disease is a serious threat to global wheat production. Wheat blast is caused by a distinct, exceptionally diverse lineage of the fungus causing rice blast disease. To understand genetic diversity in wheat-infecting strains, we report a near-finished reference genome of a recent field isolate generated using long read sequencing and a novel scaffolding approach with long-distance paired genomic sequences. The genome assemblage includes seven core chromosomes and sequences from a dispensable mini-chromosome that harbors effector genes normally found on the ends of core chromosomes in other strains. No mini-chromosomes were observed in an early field strain, and two mini-chromosomes from another field isolate each contain different effector homologous genes and core chromosome end sequences. The mini-chromosome is highly repetitive and is enriched in transposons occurring most frequently at core chromosome ends. Additionally, transposons in mini-chromosomes lack the characteristic signature for inactivation by repeat-induced point (RIP) mutation genome defenses. Our results, collectively, indicate that dispensable mini-chromosomes and non-dispensable core chromosomes undergo divergent evolutionary trajectories, and mini-chromosomes and core chromosome ends are coupled as a mobile, fast-evolving effector compartment in the wheat pathogen genome.
Oaks are an important part of our natural and cultural heritage. Not only are they ubiquitous in our most common landscapes1 but they have also supplied human societies with invaluable services, including food and shelter, since prehistoric times2. With 450 species spread throughout Asia, Europe and America3, oaks constitute a critical global renewable resource. The longevity of oaks (several hundred years) probably underlies their emblematic cultural and historical importance. Such long-lived sessile organisms must persist in the face of a wide range of abiotic and biotic threats over their lifespans. We investigated the genomic features associated with such a long lifespan by sequencing, assembling and annotating the oak genome. We then used the growing number of whole-genome sequences for plants (including tree and herbaceous species) to investigate the parallel evolution of genomic characteristics potentially underpinning tree longevity. A further consequence of the long lifespan of trees is their accumulation of somatic mutations during mitotic divisions of stem cells present in the shoot apical meristems. Empirical4 and modelling5 approaches have shown that intra-organismal genetic heterogeneity can be selected for6and provides direct fitness benefits in the arms race with short-lived pests and pathogens through a patchwork of intra-organismal phenotypes7. However, there is no clear proof that large-statured trees consist of a genetic mosaic of clonally distinct cell lineages within and between branches. Through this case study of oak, we demonstrate the accumulation and transmission of somatic mutations and the expansion of disease-resistance gene families in trees.
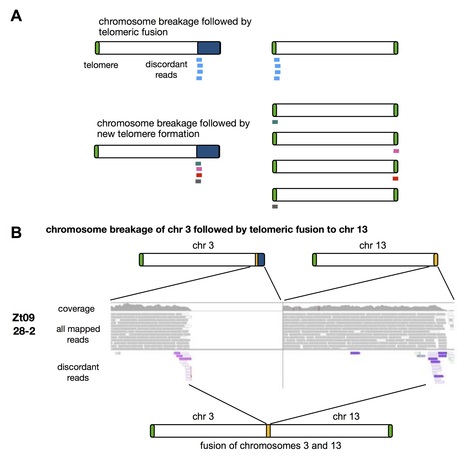
The haploid genome of the pathogenic fungus Zymoseptoria tritici is contained on “core” and “accessory” chromosomes. While 13 core chromosomes are found in all strains, as many as eight accessory chromosomes show presence/absence variation and rearrangements among field isolates. We investigated chromosome stability using experimental evolution, karyotyping and genome sequencing. We report extremely high and variable rates of accessory chromosome loss during mitotic propagation in vitro and in planta. Spontaneous chromosome loss was observed in 2 to >50 % of cells during four weeks of incubation. Similar rates of chromosome loss in the closely related Z. ardabiliae suggest that this extreme chromosome dynamic is a conserved phenomenon in the genus. Elevating the incubation temperature greatly increases instability of accessory and even core chromosomes, causing severe rearrangements involving telomere fusion and chromosome breakage. Chromosome losses do not impact the fitness of Z. tritici in vitro, but some lead to increased virulence suggesting an adaptive role of this extraordinary chromosome instability.
|
Newly emerged wheat blast disease is a serious threat to global wheat production. Wheat blast is caused by a distinct, exceptionally diverse lineage of the fungus causing rice blast disease. Through sequencing a recent field isolate, we report a reference genome that includes seven core chromosomes and mini-chromosome sequences that harbor effector genes normally found on ends of core chromosomes in other strains. No mini-chromosomes were observed in an early field strain, and at least two from another isolate each contain different effector genes and core chromosome end sequences. The mini-chromosome is enriched in transposons occurring most frequently at core chromosome ends. Additionally, transposons in mini-chromosomes lack the characteristic signature for inactivation by repeat-induced point (RIP) mutation genome defenses. Our results, collectively, indicate that dispensable mini-chromosomes and core chromosomes undergo divergent evolutionary trajectories, and mini-chromosomes and core chromosome ends are coupled as a mobile, fast-evolving effector compartment in the wheat pathogen genome.
Phytopathogen genomes are under constant pressure to change, as pathogens are locked in an evolutionary arms race with their hosts, where pathogens evolve effector genes to manipulate their hosts, while the hosts evolve immune components to recognize the products of these genes. Colletotrichum higginsianum (Ch), a fungal pathogen with no known sexual morph, infects Brassicaceae plants including Arabidopsis thaliana. Previous studies revealed that Ch differs in its virulence towards various A. thaliana ecotypes, indicating the existence of coevolutionary selective pressures. However, between-strain genomic variations in Ch have not been studied. Here, we sequenced and assembled the genome of a Ch strain, resulting in a highly contiguous genome assembly, which was compared to the chromosome-level genome assembly of another strain to identify genomic variations between strains. We found that the two closely related strains vary in terms of large-scale rearrangements, the existence of strain-specific regions, and effector candidate gene sets and that these variations are frequently associated with transposable elements (TEs). Ch has a compartmentalized genome consisting of gene-sparse, TE-dense regions with more effector candidate genes and gene-dense, TE-sparse regions harboring conserved genes. Additionally, analysis of the conservation patterns and syntenic regions of effector candidate genes indicated that the two strains vary in their effector candidate gene sets because of de novo evolution, horizontal gene transfer, or gene loss after divergence. Our results reveal mechanisms for generating genomic diversity in this asexual pathogen, which are important for understanding its adaption to hosts.
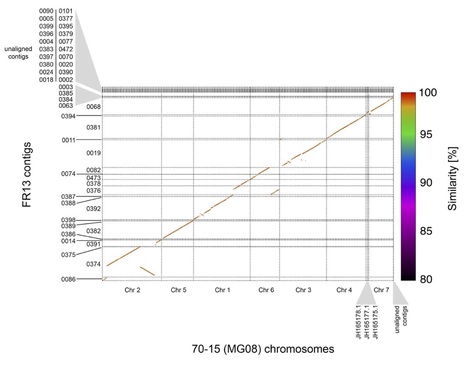
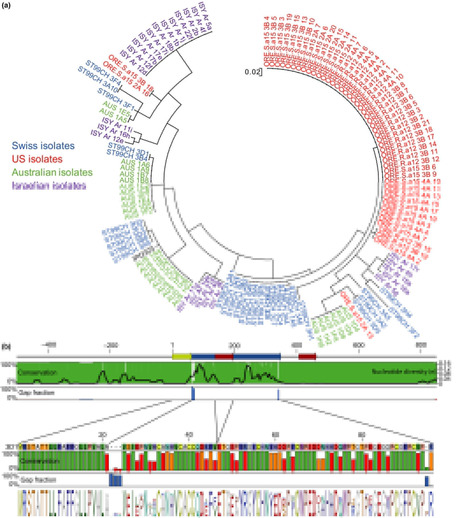
We report long-range sequencing of eight isolates of Magnaporthe oryzae(Syn. Pyricularia oryzae) from wheat, rice, foxtail millet and goosegrass using nanopore MinION. Our aim is to obtain chromosome-level genome assemblies that are freely available for public access to be scrutinized for genome rearrangements and structural variation. Magnaporthe oryzae(Syn. Pyricularia oryzae) is a notorious fungus known for causing blast disease on rice and wheat with devastating effect on grain yield. M. oryzaehost range includes several other cereal crops such as oat, finger millet and foxtail millet as well as wild grasses. M. oryzaeis found all over the world wherever warm temperature and high humidity are common. Although lineages of M. oryzaetend to be adapted to a particular host, this pathogen can shift from one host to another when the conditions permit (Couch et al.2005). We hypothesize that structural variation contributes to adaptation to new hosts and environmental conditions following, for example, the model proposed by Chuma et al.(2011). To generate the genomics data that would enable such analyses, we sequenced eight isolates of M. oryzaefrom four different hosts using long-range nanonpore MinION with the aim of providing chromosome-level genome assemblies that are freely available for public access.
- Cultivar‐strain specificity in the wheat–Zymoseptoria tritici pathosystem determines the infection outcome and is controlled by resistance genes on the host side, many of which have been identified. On the pathogen side, however, the molecular determinants of specificity remain largely unknown.
- We used genetic mapping, targeted gene disruption and allele swapping to characterise the recognition of the new avirulence factor Avr3D1. We then combined population genetic and comparative genomic analyses to characterise the evolutionary trajectory of Avr3D1.
- Avr3D1 is specifically recognised by wheat cultivars harbouring the Stb7resistance gene, triggering a strong defence response without preventing pathogen infection and reproduction. Avr3D1 resides in a cluster of putative effector genes located in a genome region populated by independent transposable element insertions. The gene was present in all 132 investigated strains and is highly polymorphic, with 30 different protein variants identified. We demonstrated that specific amino acid substitutions in Avr3D1 led to evasion of recognition.
- These results demonstrate that quantitative resistance and gene‐for‐gene interactions are not mutually exclusive. Localising avirulence genes in highly plastic genomic regions probably facilitates accelerated evolution that enables escape from recognition by resistance proteins.
- Colletotrichum lentis causes anthracnose, which is a serious disease on lentil and can account for up to 70% crop loss. Two pathogenic races, 0 and 1, have been described in the C. lentis population from lentil.
- To unravel the genetic control of virulence, an isolate of the virulent race 0 was sequenced at 1481‐fold genomic coverage. The 56.10‐Mb genome assembly consists of 50 scaffolds with N50 scaffold length of 4.89 Mb. A total of 11 436 protein‐coding gene models was predicted in the genome with 237 coding candidate effectors, 43 secondary metabolite biosynthetic enzymes and 229 carbohydrate‐active enzymes (CAZymes), suggesting a contraction of the virulence gene repertoire in C. lentis.
- Scaffolds were assigned to 10 core and two minichromosomes using a population (race 0 × race 1, n = 94 progeny isolates) sequencing‐based, high‐density (14 312 single nucleotide polymorphisms) genetic map. Composite interval mapping revealed a single quantitative trait locus (QTL), qClVIR‐11, located on minichromosome 11, explaining 85% of the variability in virulence of the C. lentis population. The QTL covers a physical distance of 0.84 Mb with 98 genes, including seven candidate effector and two secondary metabolite genes.
- Taken together, the study provides genetic and physical evidence for the existence of a minichromosome controlling the C. lentis virulence on lentil.
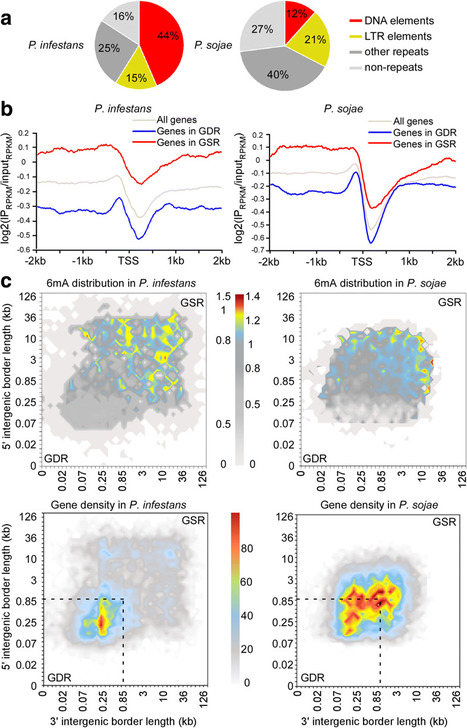
Background. Filamentous plant pathogen genomes often display a bipartite architecture with gene-sparse, repeat-rich compartments serving as a cradle for adaptive evolution. The extent to which this two-speed genome architecture is associated with genome-wide DNA modifications is unknown. Results. We show that the oomycetes Phytophthora infestans and Phytophthora sojaepossess functional adenine N6-methylation (6mA) methyltransferases that modulate patterns of 6mA marks across the genome. In contrast, 5-methylcytosine could not be detected in these species. Methylated DNA IP sequencing (MeDIP-seq) of each species reveals 6mA is depleted around the transcription start sites (TSSs) and is associated with lowly expressed genes, particularly transposable elements. Genes occupying the gene-sparse regions have higher levels of 6mA in both genomes, possibly implicating the methylome in adaptive evolution. All six putative adenine methyltransferases from P. infestans and P. sojae, except PsDAMT2, display robust enzymatic activities. Surprisingly, single knockouts in P. sojaesignificantly reduce in vivo 6mA levels, indicating that the three enzymes are not fully redundant. MeDIP-seq of the psdamt3 mutant reveals uneven 6mA methylation reduction across genes, suggesting that PsDAMT3 may have a preference for gene body methylation after the TSS. Furthermore, transposable elements such as DNA elements are more active in the psdamt3mutant. A large number of genes, particularly those from the adaptive genomic compartment, are differentially expressed.
Conclusions. Our findings provide evidence that 6mA modification is potentially an epigenetic mark in Phytophthora genomes, and complex patterns of 6mA methylation may be associated with adaptive evolution in these important plant pathogens.
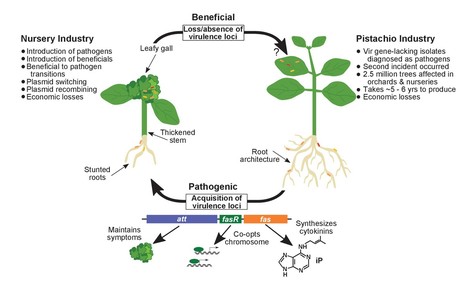
Understanding how bacteria affect plant health is crucial for developing sustainable crop production systems. We coupled ecological sampling and genome sequencing to characterize the population genetic history of Rhodococcus and the distribution patterns of virulence plasmids in isolates from nurseries. Analysis of chromosome sequences shows that plants host multiple lineages of Rhodococcus, and suggested that these bacteria are transmitted due to independent introductions, reservoir populations, and point source outbreaks. We demonstrate that isolates lacking virulence genes promote beneficial plant growth, and that the acquisition of a virulence plasmid is sufficient to transition beneficial symbionts to phytopathogens. This evolutionary transition, along with the distribution patterns of plasmids, reveals the impact of horizontal gene transfer in rapidly generating new pathogenic lineages and provides an alternative explanation for pathogen transmission patterns. Results also uncovered a misdiagnosed epidemic that implicated beneficial Rhodococcus bacteria as pathogens of pistachio. The misdiagnosis perpetuated the unnecessary removal of trees and exacerbated economic losses.
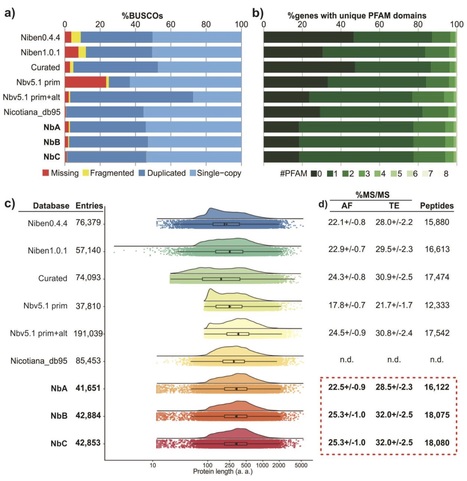
Nicotiana benthamiana is an important model organism and representative of the Solanaceae (Nightshade) family. N. benthamiana has a complex ancient allopolyploid genome with 19 chromosomes, and an estimated genome size of 3.1Gb. Several draft assemblies of the N. benthamiana genome have been generated, however, many of the gene-models in these draft assemblies appear incorrect. Here we present a nearly non-redundant database of improved N. benthamiana gene-models based on gene annotations from well-annotated genomes in the Nicotiana genus. We show that the new predicted proteome is more complete than the previous proteomes and more sensitive and accurate in proteomics applications, while maintaining a reasonable low gene number (~43,000). As a proof-of-concept we use this proteome to compare the leaf extracellular (apoplastic) proteome to a total extract of leaves. Several gene families are more abundant in the apoplast. For one of these apoplastic protein families, the subtilases, we present a phylogenetic analysis illustrating the utility of this database. Besides proteome annotation, this database will aid the research community with improved target gene selection for genome editing and off-target prediction for gene silencing.
What did you find? We studied two different races of the Irish potato famine pathogen, and we discovered that the difference invirulence between these races could not be ascribed to a genetic difference but rather to a difference in the expression of the underlying virulence gene. This adds to our knowledge of how this important scourge on world agriculture evolves to evade plant immunity. Why is this work important? As our colleague Mark Gijzen tweeted, “is this a rare and unusual curiosity or another example of a widespread biological phenomenon?” Indeed, there are few other examples in related plant pathogens, including the soybean root rot pathogen that Mark studies. This finding has far reaching implications. It indicates that these pathogens can evolve even more rapidly than anticipated thus counteracting the efforts of plant breeders to deploy disease resistant crops. Are potato varieties resistant to the pathogen available? Yes, there are. But there are several examples of potato cultivars that were initially resistant to late blight when farmers started to grow them, but succumbed to the disease a few years later. The ability to switch on and off virulence genes such as we found in this research may partly explain why the pathogen is so effective at overcoming the plants defense barriers. What is currently done to control the disease? Susceptible potato cultivars must be protected by repeated applications of fungicides. If left unchecked, the disease will destroy the leaves and stems in a matter of days as in the pictured trial plot of potato varieties in the highlands of Peru. Is chemical protection the only way to control late blight? In nature, there are wild relatives of the cultivated potato and many of them can withstand the disease (see image of potato variety field trial). Breeders identify the genes in these plants and introduce them to cultivated potato through crosses or genetic transformation. How did you put this project together? We studied an Andean lineage of the Irish potato famine pathogen known as EC-1 so the project had an international flavor from day one. Ours was a wide reaching multinational collaboration bringing together scientists based in the UK, Japan, Netherlands, USA, Philippines, and Peru. It’s how science often goes on these days. Experts from all over the world team up to solve problems, make new discoveries and advance our knowledge. Anything you would have done differently? DNA sequencing technology develops so fast that by the time the paper gets published you wish you could apply a different method. It also takes more time to analyze the data, write up the paper etc. than to generate the sequence data. This can be frustrating. You posted the paper in bioRxiv before submission. Why? Why not? Posting the article on bioRxiv enabled us to share our findings with our colleagues and hear about it from the community as soon as possible. The tweet by Mark Gijzen we referred to above is an example of such feedback. Posting a preprint relieves some of the delays associated with publishing. It’s a liberating feeling to finish writing up a paper and immediately share it with anyone who’s interested. Authors Dr. Vivianne Vleeshouwers is assistant professor in Wageningen University & Research, the Netherlands. Her research is dedicated to understand the molecular interaction between the potato late blight pathogen Phytophthora infestans and potato, and exploit this knowledge to achieve a better and more durable disease resistance. Dr. Hannele Lindqvist-Kreuze works as a Molecular Breeder at the International Potato Center (CIP) in Lima, Peru. Her current work focuses on the discovery and application of molecular markers in the potato and sweet potato breeding programs of CIP. She describes her work as a Haiku: Searching for Hidden Patterns, Coded in the DNA, Unknowingly selected. Dr. Sophien Kamoun is a Senior Scientist at The Sainsbury Laboratory and a Professor of Biology at the University of East Anglia in Norwich, UK. He studies the interactions between plants and filamentous pathogens, notably the Irish potato famine pathogen and the rice and wheat blast fungus. He’s known for saying “Don’t bet against the pathogen.”
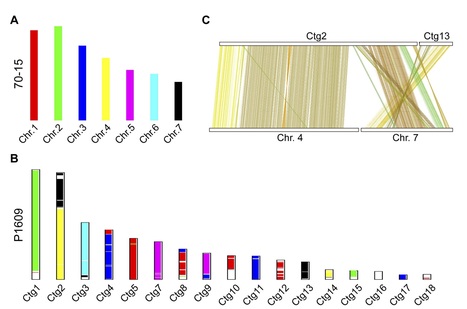
Backgrounds: Pyricularia is a multispecies complex that could infect and cause severe blast disease on diverse hosts, including rice, wheat and many other grasses. Although the genome size of this fungal complex is small [~40 Mbp for Pyricularia oryzae (syn. Magnaporthe oryzae), and ~45 Mbp for P. grisea], the genome plasticity allows the fungus to jump and adapt to new hosts. Therefore, deciphering the genome basis of individual species could facilitate the evolutionary and genetic study of this fungus. However, except for the P. oryzae subgroup, many other species isolated from diverse hosts, such as the Pennisetum grasses, remain largely uncovered genetically. Results: Here, we report the genome sequence of a pyriform-shaped fungal strain P. penniseti P1609 isolated from a Pennisetum grass (JUJUNCAO) using PacBio SMRT sequencing technology. We performed a phylogenomic analysis of 28 Magnaporthales species and 5 non-Magnaporthales species and addressed P1609 into a Pyricularia subclade that is distant from P. oryzae. Comparative genomic analysis revealed that the pathogenicity-related gene repertoires were fairly different between P1609 and the P. oryzae strain 70-15, including the cloned avirulence genes, other putative secreted proteins, as well as some other predicted Pathogen-Host Interaction (PHI) genes. Genomic sequence comparison also identified many genomic rearrangements. Conclusion: Taken together, our results suggested that the genomic sequence of the P. penniseti P1609 could be a useful resource for the genetic study of the Pennisetum-infecting Pyricularia species.
Gene-for-gene immunity between plants and host-adapted pathogens is often linked to population-level diversification of immune receptors encoded by disease resistance (R) genes. The complex barley (Hordeum vulgare L.) R gene locus Mildew Locus A (Mla) provides isolate-specific resistance against the powdery mildew fungus Blumeria graminis f. sp. hordei (Bgh) and has been introgressed into modern barley cultivars from diverse germplasms, including the wild relative H. spontaneum. Known Mla disease resistance specificities to Bgh appear to encode allelic variants of the R Gene Homolog 1 (RGH1) family of nucleotide-binding domain and leucine-rich repeat (NLR) proteins. To gain insights into Mla diversity in wild barley populations, we here sequenced and assembled the transcriptomes of 50 accessions of H. spontaneum representing nine populations distributed throughout the Fertile Crescent. The assembled Mla transcripts exhibited rich sequence diversity, which is linked neither to geographic origin nor population structure. Mla transcripts in the tested H. spontaneum accessions could be grouped into two similar-sized subfamilies based on two major N-terminal coiled-coil signaling domains that are both capable of eliciting cell death. The presence of positively selected sites, located mainly in the C-terminal leucine-rich repeats of both MLA subfamilies, together with the fact that both coiled-coil signaling domains mediate cell death, implies that the two subfamilies are actively maintained in the host population. Unexpectedly, known MLA receptor variants that confer Bgh resistance belong exclusively to one subfamily. Thus, signaling domain divergence, potentially to distinct pathogen populations, is an evolutionary signature of functional diversification of an immune receptor.
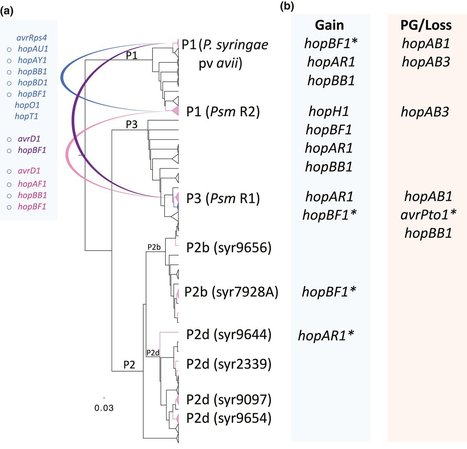
- Genome‐wide analyses of the effector‐ and toxin‐encoding genes were used to examine the phylogenetics and evolution of pathogenicity amongst diverse strains of Pseudomonas syringae causing bacterial canker of cherry (Prunus avium), including pathovars P. syringae pv morsprunorum (Psm) races 1 and 2, P. syringae pv syringae(Pss) and P. syringae pv avii.
- Phylogenetic analyses revealed Psm races and P. syringae pv avii clades were distinct and were each monophyletic, whereas cherry‐pathogenic strains of Pss were interspersed amongst strains from other host species.
- A maximum likelihood approach was used to predict effectors associated with pathogenicity on cherry. Pss possesses a smaller repertoire of type III effectors but has more toxin biosynthesis clusters than Psm and P. syringae pv avii. Evolution of cherry pathogenicity was correlated with gain of genes such as hopAR1 and hopBB1through putative phage transfer and horizontal transfer respectively. By contrast, loss of the avrPto/hopAB redundant effector group was observed in cherry‐pathogenic clades. Ectopic expression of hopAB and hopC1 triggered the hypersensitive reaction in cherry leaves, confirming computational predictions.
- Cherry canker provides a fascinating example of convergent evolution of pathogenicity that is explained by the mix of effector and toxin repertoires acting on a common host.
|