 Your new post is loading...
Your new post is loading...
The Vaccinology Club of the French Society of Immunology, created in 2001 by Claude Leclerc, will hold its next conference on vaccinology, the 16-17th of January 2020 at the Institut Pasteur in Lille. The scientific committee is composed of top French key opinion leaders on vaccinology. Many aspects of vaccinology will thus be addressed with sessions devoted i) to advances in fundamental research, ii) the development of new vaccine approaches and new methods for monitoring the effectiveness of vaccines such as systems vaccinology, iii) and the latest results of clinical trials. You will note in the list of speakers who have already agreed to participate, the participation of prestigious International speakers and French vaccinology stakeholders. One of the club’s roles is also to help raising awareness of the work of young scientists or clinical trial investigators, and our symposium will give them a prominent place in oral abstract and poster sessions. We hope to be able to create a new generation of committed people interested in vaccine development and application. The success of this conference has been largely based on your participation the financial support of academic and industrial sponsors.
A vaccine to fight Lassa fever
Thousands of people in Western Africa succumb to Lassa fever each year. Lassa virus infections can in some cases respond to antiviral treatment, but there is no vaccine. Mateo et al. tested three different viral-vectored Lassa virus vaccines in a nonhuman primate model. They compared transcriptomic and proteomic signatures as well as B cell and T cell responses after vaccination. One vaccine using a measles virus vector with Lassa virus glycoprotein and nucleoprotein provided almost sterilizing protection upon a lethal Lassa virus challenge. This vaccine will soon be tested in humans and has the potential to make a major impact on the incidence of Lassa fever.
BRISBANE, Calif.--(BUSINESS WIRE)--Sep. 13, 2019-- Aimmune Therapeutics, Inc. (Nasdaq:AIMT), a biopharmaceutical company developing treatments for potentially life-threatening food allergies, today announced that the Allergenic Products Advisory Committee (APAC) convened by the U.S. Food and Drug Administration (FDA) voted to support the use of AR101 (proposed trade name PALFORZIA™) in children and teens with peanut allergy. PALFORZIA is a complex, biologic oral immunotherapy (OIT) candidate designed to reduce the incidence and severity of allergic reactions, including anaphylaxis, after accidental exposure to peanut in patients aged 4 through 17 years with a confirmed diagnosis of peanut allergy.
The APAC voted 7 to 2 that the efficacy data and 8 to 1 that the safety data, in conjunction with additional safeguards, are adequate to support the use of PALFORZIA.
Despite efforts made to develop efficient preventive strategies, influenza A virus (IAV) infections continue to cause serious clinical and economic problems. Current licensed human vaccines are mainly inactivated whole virus particles or split-virion administrated via the parenteral route. These vaccines provide incomplete protection against IAV in high-risk groups and are poorly/not effective faced with constant antigenic drift/shift occurring in circulating strains. Advances in mucosal vaccinology and in the understanding of the protective anti-influenza immune mechanisms suggest that intranasal immunization is a promising strategy to fight against IAV. To date, human mucosal anti-influenza vaccines consist of live attenuated strains administrated intranasally, which elicit higher local humoral and cellular immune responses than conventional parenteral vaccines. However, because of inconsistent protective efficacy and safety concerns regarding the use of live viral strains, new vaccine candidates are urgently needed. To prime and induce potent and long-lived protective immune responses, mucosal vaccine formulations need to ensure the immunoavailability and the immunostimulating capacity of the vaccine antigen(s) at the mucosal surfaces, while being minimally reactogenic/toxic. The purpose of this review is to compile innovative delivery/adjuvant systems tested for intranasal administration of inactivated influenza vaccines, including micro/nanosized particulate carrier
The NIH has awarded $2.3M for a pilot project, led by Boston Children's Hospital, to develop a vaccine that would protect people with opioid use disorder against an accidental fentanyl overdose.
The V1V2 region of HIV Env is an important vaccine target. In comparative immunogenicity
studies in macaques, Hessell et al. reveal that a cocktail of multiple V1V2-scaffold immunogens readily induces durable, polyfunctional V1V2-specific antibodies with antiviral activities that are associated...
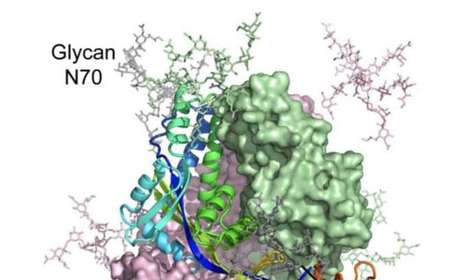
A nanoparticle Epstein-Barr virus glycoprotein gH/gL vaccine elicits antibodies that mediate cell type–independent protection from infection.
|
Summary Although epidemiological observations, IgG passive transfer studies and experimental infections in humans all support the feasibility of developing highly effective malaria vaccines, the pr...
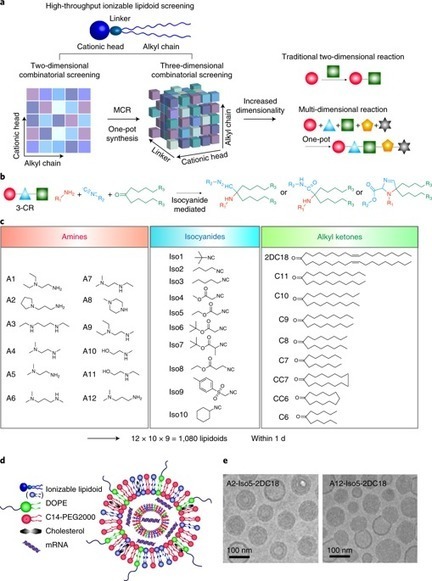
Therapeutic messenger RNA vaccines enable delivery of whole antigens, which can be advantageous over peptide vaccines. However, optimal efficacy requires both intracellular delivery, to allow antigen translation, and appropriate immune activation. Here, we developed a combinatorial library of ionizable lipid-like materials to identify mRNA delivery vehicles that facilitate mRNA delivery in vivo and provide potent and specific immune activation. Using a three-dimensional multi-component reaction system, we synthesized and evaluated the vaccine potential of over 1,000 lipid formulations. The top candidate formulations induced a robust immune response, and were able to inhibit tumor growth and prolong survival in melanoma and human papillomavirus E7 in vivo tumor models. The top-performing lipids share a common structure: an unsaturated lipid tail, a dihydroimidazole linker and cyclic amine head groups. These formulations induce antigen-presenting cell maturation via the intracellular stimulator of interferon genes (STING) pathway, rather than through Toll-like receptors, and result in limited systemic cytokine expression and enhanced anti-tumor efficacy. Combinatorial development of heterocyclic lipidoids yields efficient mRNA vaccine vehicles that activate immune cells via the STING pathway.
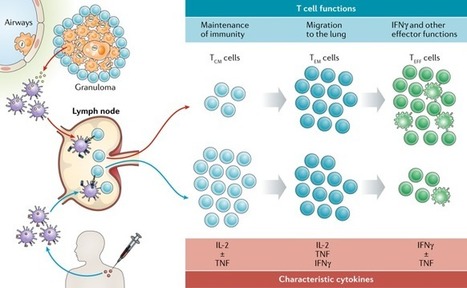
Tuberculosis (TB) vaccine research has reached a unique point in time. Breakthrough findings in both the basic immunology of Mycobacterium tuberculosis infection and the clinical development of TB vaccines suggest, for the first time since the discovery of the Mycobacterium bovis bacillus Calmette–Guérin (BCG) vaccine more than a century ago, that a novel, efficacious TB vaccine is imminent. Here, we review recent data in the light of our current understanding of the immunology of TB infection and discuss the identification of biomarkers for vaccine efficacy and the next steps in the quest for an efficacious vaccine that can control the global TB epidemic.
KEY POINTS
Human CD4 T cells have limited cross-reactive memory to avian H7 proteins.
CD4 T cell help can be provided to B cells with novel H7/H3 chimeric vaccines.
Abstract
Avian influenza vaccines exhibit poor immunogenicity in humans. We hypothesized that one factor underlying weak B cell responses was sequence divergence between avian and seasonal influenza hemagglutinin proteins, thus limiting the availability of adequate CD4 T cell help. To test this, a novel chimeric hemagglutinin protein (cH7/3) was derived, comprised of the stem domain from seasonal H3 hemagglutinin and the head domain from avian H7. Immunological memory to seasonal influenza was established in mice, through strategies that included seasonal inactivated vaccines, Flumist, and synthetic peptides derived from the H3 stalk domain. After establishment of memory, mice were vaccinated with H7 or cH7/3 protein. The cH7/3 Ag was able to recall H3-specific CD4 T cells, and this potentiated CD4 T cell response was associated with enhanced early germinal center response and rapid elicitation of Abs to H7, including Abs specific for the H7 head domain. These results suggest that in pandemic situations, inclusion of CD4 T cell epitopes from seasonal viruses have the potential to overcome the poor immunogenicity of avian vaccines by helping B cells and conferring greater subtype-specific Ab response to viral HA.
An experimental vaccine against respiratory syncytial virus (RSV), one of the leading causes of infectious disease deaths in infants, has shown early promise in a Phase 1 human clinical trial. A team of researchers, including ...
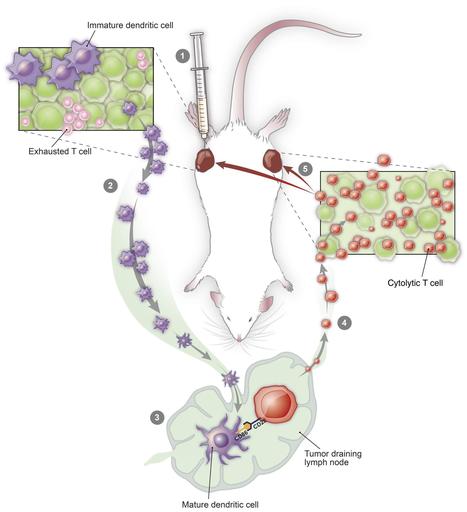
Research ArticleOncology Free access | 10.1172/JCI128562 In situ vaccination with defined factors overcomes T cell exhaustion in distant tumors Danny N. Khalil,1,2,3,4 Nathan Suek,1 Luis Felipe Campesato,1 Sadna Budhu,1 David Redmond,1 Robert M. Samstein,5 Chirag Krishna,6 Katherine S. Panageas,7 Marinela Capanu,7 Sean Houghton,1 Daniel Hirschhorn,1 Roberta Zappasodi,1,2 Rachel Giese,1,8 Billel Gasmi,9 Michael Schneider,1 Aditi Gupta,1 James J. Harding,3,4 John Alec Moral,8 Vinod P. Balachandran,2,10 Jedd D. Wolchok,1,2,3,4 and Taha Merghoub1,2,3,4 First published July 22, 2019 - More info Abstract Irreversible T cell exhaustion limits the efficacy of programmed cell death 1 (PD-1) blockade. We observed that dual CD40-TLR4 stimulation within a single tumor restored PD-1 sensitivity and that this regimen triggered a systemic tumor-specific CD8+ T cell response. This approach effectively treated established tumors in diverse syngeneic cancer models, and the systemic effect was dependent on the injected tumor, indicating that treated tumors were converted into necessary components of this therapy. Strikingly, this approach was associated with the absence of exhausted PD-1hi T cells in treated and distant tumors, while sparing the intervening draining lymph node and spleen. Furthermore, patients with transcription changes like those induced by this therapy experienced improved progression-free survival with anti–PD-1 treatment. Dual CD40-TLR4 activation within a single tumor is thus an approach for overcoming resistance to PD-1 blockade that is unique in its ability to cause the loss of exhausted T cells within tumors while sparing nonmalignant tissues. Graphical Abstract Introduction Immune checkpoint blockade (ICB) has improved outcomes for patients with diverse cancers, yet most patients with common cancers still do not show a clinical response. Features of malignant cells (1) as well as the immune infiltrate (2) play an important role in mediating resistance. In the latter category, T cell exhaustion acts as a key barrier to effective immunotherapy (3). Here, we propose that antigen-presenting cell (APC) activation status within a single tumor can impact tumor-associated T cell exhaustion systemically. We sought to overcome the hurdle imposed by T cell exhaustion through local APC activation using low-dose intratumoral therapy. We hypothesized that such an approach would augment the activity of programmed cell death 1 (PD-1) blockade for 2 reasons. First, because activating APCs exposed to tumor antigens may prime new waves of tumor-specific T cells; and second, because PD-1 has been shown to regulate CD28 signaling in T cells (4, 5), which is triggered by the costimulatory ligands CD80 and CD86 on activated APCs (6). Using syngeneic murine models, we identified an intratumoral treatment approach using dual CD40-TLR4 stimulation that can overcome tumor-associated T cell exhaustion. Remarkably, this approach eliminated phenotypically exhausted CD8+ T cells at both treated and distant (so-called abscopal; refs. 7, 8) tumors, while sparing lymphoid organs such as the intervening draining lymph node (DLN) and spleen. Results Defined factors control local and distant tumors. To investigate the possibility of a causal relationship between APC activation, particularly CD86 expression, and an antitumor immune response, we used monophosphoryl lipid A (MPL), a low-toxicity TLR4 agonist that induces APC activation in human and murine APCs (9, 10). We used C57BL/6 bone marrow–derived APCs to determine whether MPL induces CD86 expression and phagocytosis in vitro in the CD11chi cell population (Figure 1A). We next examined whether these effects were maintained in tumors in vivo using the syngeneic B16F10 melanoma model. We found that MPL retains the ability to augment phagocytosis, as it induced the uptake of intratumorally implanted latex particles (11) by endogenous CD11chi phagocytic cells (Figure 1B). However, MPL did not retain the ability to activate APCs in the tumor microenvironment (Figure 1B). Given the potent role that CD40 can play in APC activation (12) and its ability to sensitize DCs to TLR stimulation (13), we tested the effect of an agonistic CD40 mAb on APC activation in vivo when administered intratumorally. We found that anti-CD40 induced the activation of CD11chi cells following intratumoral administration (Figure 1B) in this system. Figure 1 Agents that promote phagocytosis and APC activation, but not direct tumor cell lysis, control local and distant tumors. (A) Bone marrow–derived myeloid cells were treated with MPL or vehicle for 16 hours and incubated with FITC-labeled latex beads. Flow cytometry was performed to determine the fraction of CD11chi cells that phagocytosed FITC-labeled beads and the median fluorescence intensity (MFI) of CD86 expression (n = 3 – 5/group). (B) C57BL/6 mice were implanted intradermally with 5 × 105 B16F10 cells. On day 8, FITC-labeled latex beads were coinjected intratumorally with vehicle, MPL, or anti-CD40. Twenty-four hours later, the CD11chi cell population was analyzed in tumors (left) for phagocytosis (n = 5/group) and in DLNs (right) for CD86 expression (n = 4/group). (C) Treatment schedule: intratumoral biweekly treatments, with or without intraperitoneal anti–PD-1, were started once bilateral tumors were established; treatment was continued for 4 weeks. (D) Individual growth curves of treated and distant tumors in animals treated with MPL and anti-CD40 (n = 10/group). (E) Average tumor growth curves comparing MPL and anti-CD40 with constituent monotherapies (n = 10/group). (F) Viability of B16F10 cells treated in vitro with MPL, anti-CD40, or gemcitabine for 72 hours. (G) Growth of treated and distant tumors upon addition of anti–PD-1 (n = 10/group). *P ≤ 0.0, **P ≤ 0.01, ***P ≤ 0.001, and ****P ≤ 0.0001, by unpaired, 2-tailed Student’s t test. Hypothesizing that intratumoral administration of agents mediating phagocytosis and APC activation would trigger a systemic antitumor immune response, we used a bilateral tumor approach (Figure 1C). This allowed us to distinguish the impact of therapy on the treated tumor from that on the distant tumor in animals bearing established, concurrently implanted tumors. We found that the combination of MPL and anti-CD40 eradicated or delayed the growth of treated and distant tumors, respectively (Figure 1D), and that this combination conferred greater antitumor activity than did either anti-CD40 or MPL monotherapy at both the treated and distant tumors (Figure 1E). To assess the possibility that MPL or anti-CD40 was directly cytotoxic, we asked whether these agents directly affect B16F10 viability in vitro. In contrast to oncolytic agents used for in situ vaccination (14), we found that neither MPL nor anti-CD40 demonstrated direct cytolytic activity (Figure 1F). Given the potential of activated APCs to prime antitumor T cells, we next asked whether the addition of anti–PD-1 treatment would augment treatment efficacy in this PD-1–resistant (15, 16) model. We found that addition of anti–PD-1 improved tumor control at both the treated and distant tumors (Figure 1G). Treatment efficacy depends on BATF3+ DCs and CD8+ T cells. To understand the mechanism through which the anti-CD40, MPL, and anti–PD-1 (CMP) regimen mediates antitumor activity, we analyzed distant tumors after 1 week of treatment and found that CMP-treated, but not isotype-treated, animals developed lymphocytic infiltrates deep within the tumor (Figure 2A). This corresponded with an increased fraction of CD8+ T cells and greater proliferation within this population (Figure 2A). The distant tumors continued to be enriched for CD8+ T cells after 3 weeks of treatment, and this enrichment became more pronounced by 6 weeks (Figure 2B), at which point only the treated animals were alive. Figure 2 CMP combination therapy augments APC activation and nodal accumulation followed by a systemic CD8+ T cell response. (A) Using the bilateral tumor model, distant tumors from isotype- (Control) and CMP-treated (Trx) animals were assessed by H&E staining after 1 week of treatment (scale bars: 50 μm) and by flow cytometry to quantify CD8+ T cell infiltrates and the fraction of this cell population expressing Ki67 (n = 4/group). (B) Distant tumors were analyzed by immunofluorescence (IF) at 3 and 6 weeks for CD4 (green), FoxP3 (yellow), and CD8 (red) cell populations (scale bars: 50 μm). Quantification of the CD8+ fraction of DAPI+ cells in IF images (n = 3–10/group). N/A, no remaining live animals. (C) Growth of treated and distant tumors from WT or Rag1–/– C57BL/6 animals (n = 10/group). (D) Growth of treated and distant tumors from mice depleted of CD4+ and CD8+ T cells. Peripheral blood was collected to confirm the absence of corresponding cell populations (n = 10/group). (E) Tumor growth in mice bearing treated WT B16F10 and distant B78H10 tumors (n = 9–10/group). (F) Mice previously cured of unilateral B16F10 tumors with CMP treatment and age-matched naive controls were implanted with tumors on day 90 (n = 8–10/group). Adjacent panel shows fur depigmentation at the site of the initial cured tumor (green arrowhead) and at the site of post-treatment tumor reimplantation (red arrowhead). (G) CD86 expression in the CD11chi cell population in the treated tumor and DLN (n = 4/group). (H) Fraction of CD11chi cells among live CD45+ cells in the treated tumor and DLN (n = 4/group). (I) Tumor growth of WT or Batf3–/– C57BL/6 animals bearing B16F10 tumors treated with isotype or CMP (n = 10/group). *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, and ****P ≤ 0.0001, by unpaired, 2-tailed Student’s t test. Given the evidence of a proliferative CD8+ T cell infiltrates within abscopal tumors, we next asked whether this cell population was necessary for treatment efficacy. We found that CMP efficacy was lost in Rag1–/– mice lacking mature lymphocytes (Figure 2C) and mice depleted of CD8+, but not CD4+, T cells (Figure 2D). Given the necessity of CD8+ T cells in mediating treatment efficacy, we next used the MHC-deficient B16 cell line B78H1 (17) to confirm MHC-dependent killing of abscopal tumor cells. Mice were implanted with B16 at the treated site and B78H1 at the distant site. While local B16 tumors regressed, we observed that treatment efficacy was indeed lost at the distant B78H1 tumors (Figure 2E and Supplemental Figure 1; supplemental material available online with this article; https://doi.org/10.1172/JCI128562DS1). To assess for persistent immunity, the cured animals treated initially with CMP were reimplanted with B16F10 in the contralateral flank 90 days after treatment. All such animals fully resisted tumor reimplantation, whereas all of the naive animals developed growing tumors. Interestingly, autoimmune fur depigmentation occurred at both the site of the initial tumor and the site of tumor reimplantation, despite the absence of ongoing therapy, in a subset of animals (Figure 2F). We next examined the effect of CMP treatment on APCs to identify a cell population that may be interacting with the CD8+ T cells controlling tumor growth. We found that the CD11chi immune cell population, an important category of APCs within the tumor microenvironment (18), became activated within 3 hours of treatment and that activation persisted for 24 hours within the tumor (Figure 2G). As this occurred, we observed a delayed wave of CD11chi activation in the DLN that was evident 24 and 48 hours after treatment (Figure 2G). Strikingly, during these waves of activation, the CD11chi cell population was reduced within the tumor and concurrently accumulated within the DLN (Figure 2H). This pattern of CD11chi cell activation in the tumor followed by its activation in the DLN, together with the depletion of this cell population in the tumor and concurrent accumulation in the DLN, is consistent with nodal trafficking of activated APCs (19). Given these findings in the CD11chi cell population and the necessity of CD8+ T cells for tumor control, we asked whether BATF3+ DCs were necessary for treatment efficacy, as these DCs efficiently cross-present antigens to CD8+ T cells (20–22). Using animals deficient for BATF3+ DCs (20), we found that treatment efficacy was indeed lost in this system (Figure 2I), confirming the necessity of this DC population for treatment efficacy. CMP acts on tumor-specific T cells. We next sought to determine whether CMP induced a tumor-specific T cell response, as would be expected if BATF3+ DCs exposed to tumor antigens were priming naive CD8+ T cells. We performed an IFN-γ enzyme-linked immune absorbent spot (ELISpot) assay on CD8+ T cells sorted from treated animals 1 week after treatment and found that CMP selectively increased the response to B16F10 relative to syngeneic control tumor cells (Figure 3A). Next, we adoptively transferred Pmel-1 CD8+ T cells, specific to the melanoma differentiation antigen gp100, derived from T cell receptor–transgenic (TCR-transgenic) mice (23) into B16F10 tumor–bearing animals. As expected, we found that treatment induced proliferation in this B16F10-targeting T cell population (Figure 3B). To further assess the functional impact of CMP treatment, we used an ex vivo killing assay (24), in which CD8+ T cell cells were isolated from isotype- or CMP-treated animals. Since T cell quantities were held constant ex vivo, we were able to determine whether the cytolytic capacity of individual T cells was augmented by treatment and found that CMP enhanced the ability of individual T cells to lyse B16F10 tumor cells (Figure 3C). To determine whether CMP induces the selective expansion of endogenous T cells specific to the tumor and assess the contribution of individual treatment components, we used combinatorial encoding of MHC multimers (25). This system, capable of detecting rare endogenous T cells specific to an antigen of interest, revealed that CMP markedly expanded endogenous gp100-specific T cells that were nearly undetectable at baseline (Figure 3D). Figure 3 CMP stimulates tumor-specific CD8+ T cells. (A) B16F10 tumor–bearing mice were treated for 1 week with CMP or isotype control. CD8+ T cells were purified from spleens and restimulated with irradiated ID8 or B16F10 stimulator cells. Shown are IFN-γ+ cytotoxic T lymphocytes (CTLs) in CMP and isotype samples for both tumor cell types (n = 3–5/group). (B) One week after tumor challenge, mice bearing B16F10 tumors were adoptively transferred with 2 × 106 CellTrace Violet–labeled (CTV-labeled) Pmel-1 CD8+ T cells purified from naive TCR-transgenic mice. A single treatment with CMP or isotype was administered on day 8, and LNs were harvested on day 11 to assess proliferation. Quantification of undiluted cells is shown in the CTV dilution histograms (n = 4–5/group), which are representative of 2 experiments. (C) CD8+ T cells were purified from distant tumors of animals treated for 1 week with CMP and used for an ex vivo collagen-fibrin gel–based killing assay. CD8+ T cells were coincubated with B16 tumor cells, and tumor cell numbers were assessed using a clonogenic assay to determine the proportion of B16 cells killed and the killing constant. (D) Tumor-bearing mice were sacrificed after 1 week of CMP treatment, and DLNs were stained with H2-Db MHC class I multimers bearing peptides from the melanoma differentiation antigen gp100. Quantification of endogenous gp100-specific CD8+ T cells relative to the total CD8+ T cell population is shown in representative plots (n = 3–5/group). **P ≤ 0.01 and ****P ≤ 0.0001, by unpaired, 2-tailed Student’s t test. Having determined that CMP therapy augments the tumor-specific T cell response in a B16F10 model, we next asked whether this therapy is effective in other syngeneic models and found that it was. We observed activity in bilateral syngeneic models of colorectal, hepatocellular, and bladder cancer (Figure 4A) applying the same bilateral tumor approach used for the B16F10 tumors (Figure 1C). We next used the orthotopic, syngeneic KPC pancreatic cancer model, in which anti-CD40 monotherapy has had little to no efficacy (26). In animals bearing established KPC tumors, we found that intravenous administration was effective (without a dose increase compared with intratumoral treatment) and even induced cures in nearly half of the treated mice, whereas PD-1 monotherapy was not significantly more effective than isotype control antibody treatment (Figure 4B). Figure 4 CMP is active across tumor models but inactive in unmatched distant tumors. (A) Average growth curves of animals bearing bilateral MC38, MB49, and Hep-55.1c tumors (n = 10/group). (B) Kaplan-Meier curves show survival rates of C57BL/6 animals bearing established orthotopic KPC pancreatic tumors treated intravenously with anti-CD40 and MPL (n = 10/group). (C) Bilateral tumor model with distant B16F10 tumors and either MC38, Hep55.1c, or B16F10 (positive control) as the treated tumor (n = 10/group). *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, and ****P ≤ 0.0001, by unpaired, 2-tailed Student’s t test. Treatment efficacy is lost in unmatched bilateral syngeneic tumors. Having seen efficacy in diverse tumor models, we returned to the bilateral system, but now with unmatched syngeneic tumors at each flank (Figure 4C). Importantly, since we previously found that distant B16F10 tumors are sensitive to CMP treatment when the treated tumor is also B16F10, we used distant B16F10 tumors for all unmatched bilateral tumor experiments. This would allow us to exclude the possibility that the injected agents control distant tumors by simple diffusion and direct contact. The syngeneic tumor cell lines Hep-55.1C and MC38 (hepatocellular and colorectal carcinoma lines, respectively) were used as the treated tumors and were implanted concurrently with the distant B16F10 tumors. Consistent with tumor-specific T cell–mediated killing, these experiments revealed that when different syngeneic tumors were used as the treated tumor, distant B16F10 tumors were not controlled with CMP relative to PD-1 blockade alone (Figure 4C), indicating that the treated tumor itself is a necessary component of this therapy. CMP is associated with the absence of exhausted T cells selectively within tumors. To further characterize the systemic impact of treatment, we conducted RNA profiling of distant tumors 1 week after starting CMP treatment. We observed transcriptional changes consistent with robust T cell activity (Figure 5A). For example, granzyme K, a gene expressed in activated T cells and associated with cytolytic activity (27, 28), was the most significantly upregulated gene in distant tumors with CMP treatment versus isotype, whereas it was not significantly upregulated with anti–PD-1 alone (Figure 5A). To better understand these results, we asked whether transcriptional changes in the distant tumor at 1 week were associated with a particular T cell phenotype. Using annotated gene sets from the Molecular Signatures Database (29) defined by genes upregulated in effector versus exhausted CD8+ T cells during chronic infection (29), we found clear separation among treatment samples along the exhausted to effector spectrum. Encouragingly, samples from animals treated with PD-1 monotherapy (which is known to reinvigorate a subset of exhausted T cells) (30) appeared less exhausted than those treated with isotype control and segregated away from these samples with unsupervised clustering (Figure 5B). Strikingly, all 4 samples treated with CMP were shifted further away from an exhausted expression pattern than those treated with anti–PD-1, despite having administered the intratumoral therapy into the contralateral tumor (Figure 5B). Figure 5 CMP selectively eliminates PD-1hi T cells in tumors while sparing nontumor tissues. (A) Expression profiling of distant tumors in bilateral tumor–bearing mice after 1 week of CMP treatment (n = 4/group). Shown are transcriptional changes induced by PD-1 monotherapy or CMP therapy relative to isotype control. (B) Heatmap generated using unsupervised clustering based on annotated gene sets from the Molecular Signatures Database defined by genes upregulated in effector versus exhausted CD8+ T cells during chronic infection (FDR q value = 7.35 × 10–11). (C) Distant tumors from bilateral B16F10 tumor–bearing mice were assessed by flow cytometry to determine the fraction of PD-1hiEomeshi terminally exhausted CD8+ T cells (n = 5/group) 1 week after isotype and CMP treatment. (D) IFN-γ and granzyme B expression was quantified by flow cytometry in the distant tumors and spleens 1 week after isotype or CMP treatment (n = 5/group). (E) Changes in the PD-1hi fraction of CD8+ T cells over time (n = 4/group) in isotype- and CMP-treated animals. (F) CD8+ T cell expression of TIM3, LAG3, and 2B4 at distant tumors and spleens (n = 5/group). (G) PD-1hi fraction of CD8+ T cells in treated tumors, distant tumors, spleens, and DLNs in response to treatment with CMP and each constituent agent (n = 4/group). Representative contour plots of 2 experiments are shown for the distant tumor. *P ≤ 0.05, ** P ≤ 0.01, ***P ≤ 0.001, and ****P ≤ 0.0001, by unpaired, 2-tailed Student’s t test. Given this shift away from an exhausted CD8+ T cell expression pattern toward one associated with effector CD8+ T cells, we hypothesized that CMP treatment allows APCs exposed to tumor antigens to prime tumor-specific T cells, which then accumulate within tumors to replace existing exhausted T cells. We assessed the fraction of PD-1hiEomeshiCD8+ T cells, the population of T cells associated with terminal exhaustion and resistance to PD-1 blockade (31), and found that treatment was associated with the absence of this cell population in distant tumors (Figure 5C). Functionally, we observed that CD8+ T cells in these tumors expressed higher levels of IFN-γ and granzyme B (Figure 5D). The change in the terminally exhausted T cell population was reflected in the loss of PD-1hi T cells, which became most pronounced in treated and distant tumors 1 week after the start of CMP treatment (Figure 5E) and is consistent with enhanced cytolytic function of individual T cells sorted from CMP-treated animals’ distant tumors at 1 week (Figure 3C). We next expanded our analysis to DLNs and the spleen to further assess the loss of PD-1hiCD8+ T cells outside of tumors. Interestingly, we found no similar loss of this cell population in these lymphoid organs (Figure 5E). We next examined the expression of TIM3, LAG3, and 2B4, other cell-surface markers associated with T cell exhaustion, in distant tumors and spleens. We found that all were reduced upon CMP treatment in the tumors but were either unchanged or upregulated outside of the tumors (Figure 5F). Next, we sought to determine the contribution of constituent monotherapies to the loss of exhausted PD-1hi T cells in tumors. We found that the selective absence of exhausted T cells within tumors conferred by CMP treatment could not be recapitulated with any of the respective constituents (Figure 5G). The findings with anti–PD-1 monotherapy also confirm the lack of cross-blocking between the therapeutic (RMP 1-14) and staining (RMP 1-30) anti–PD-1 antibodies (32). Finally, given the loss of intratumoral terminally exhausted T cells with CMP treatment, we hypothesized that patients with melanoma with a baseline gene expression pattern like that induced by CMP in our melanoma model would benefit most from anti–PD-1 therapy. To address this, we analyzed publicly available RNA-Seq data (ref. 33; as described in Methods) to ask whether the gene set differentially expressed in tumors after CMP treatment (Supplemental Table 1) is associated with favorable outcomes when found in pretreatment biopsies of nivolumab-treated patients. Indeed, we found that patients with such changes at baseline went on to experience significantly improved progression-free survival with nivolumab (Figure 6A). In sum, these findings suggest a model in which treatment induces the activation of BATF3+ DCs exposed to tumor antigens which then traffic to DLNs, where they prime and expand naive tumor-specific CD8+ T cells, which in turn home to tumors, replace exhausted T cell populations, and mediate tumor regression (Figure 6B). Figure 6 Differentially expressed gene set from CMP-treated animals is associated with superior outcomes in nivolumab-treated patients. (A) Progression-free survival for patients on nivolumab from the Riaz et al. cohort (n = 51) with high versus low expression of the gene set identified from differential gene expression analysis of distant tumors from mice (n = 5) treated with CMP versus isotype at 1 week (P = 0.0032, log-rank test; HR = 0.29, 95% CI = 0.11–0.69). **P ≤ 0.01, by log-rank (Mantel-Cox) test. (B) Treatment model in which intratumoral therapy induces the activation of APCs within the tumor, which then traffic to the DLNs, where new waves of antitumor T cells are primed and gain effector function. These effector T cells then circulate systemically and ultimately accumulate in local and distant tumors to replace previously exhausted T cells that had failed to control tumor growth. Discussion Having previously described the abscopal effect in the setting of radiotherapy (34) and oncolytic viral therapy (35, 36), we now show that in situ vaccination with defined nononcolytic factors can potentiate PD-1 blockade. To our knowledge, CMP is the first treatment shown to induce the absence of terminally exhausted T cells in local and distant tumors while sparing intervening lymphoid organs. At baseline, exhaustion of antitumor T cells results from the chronic TCR stimulation of a relatively small number of T cells interacting with tumor antigens. It is tempting to speculate that by expanding this pool of T cells, CMP prevents the chronic TCR stimulation that would otherwise occur. This would potentially result in the absence of exhaustion among antitumor T cells (a population enriched within tumors), whereas exhaustion among a subset of other T cells would persist in lymphoid organs. Future studies will be needed to determine the specificity of the PD-1hi T cell populations in lymphoid organs and whether they indeed represent functionally exhausted T cells. The cause of the more robust tumor control at the treated tumor compared with the distant tumor, a finding with multiple in situ vaccines (36, 37), remains unclear and is currently being investigated in our laboratory. The use of defined factors further clarifies the minimal elements necessary to induce an abscopal effect. It has been suggested that directly inducing antigen release with radiation, cryoablation, radiofrequency ablation, oncolytic viruses, and other lytic factors is critical for initiating a systemic immune response with local modalities (38). In contrast to these approaches, CMP does not directly induce antigen release. Our results thus suggest that baseline antigen release, perhaps resulting from pretreatment turnover of malignant cells, may obviate the need for intervention to release antigen. Furthermore, oncolytic modalities are associated with the introduction of diverse damage- and pathogen-associated molecular patterns. These inflammatory molecules stimulate various pattern recognition receptors. The question of which receptors are strictly necessary for inducing a systemic immune response with such modalities had thus been left open. From a clinical perspective, CMP treatment holds significant therapeutic potential for several reasons. First, toxicity may be limited, given both the low-dose intratumoral treatment and the absence of exhausted T cells selectively within tumors. Second, each of the agents in this regimen has already been used in humans (39, 40). Third, by relying on factors with well-defined targets, CMP may avoid unanticipated off-target effects. And last, by using defined factors, CMP would reduce the likelihood of limitations associated with pathogen-based (e.g., viral and bacterial) approaches including neutralizing immunity (41). While we did not conduct a systematic study to rule out toxicity associated with treatment, no discernable toxicity was observed in the treated animals apart from fur depigmentation. Since vitiligo (an analogous effect resulting from the loss of melanocytes) has been observed in a subset of patients treated with ICB, it is possible that this adverse effect may prove to be more common in humans treated with CMP. Unlike other forms of therapeutic anticancer vaccination, in situ vaccination circumvents the need to identify and synthesize antigens specific to individual patients’ tumors. In principle, when the tumor itself is used as the source of antigen for a therapeutic vaccine, treatment can be expedited, and evolving sets of tumor antigens that arise during treatment can be targeted as they emerge. Future studies will be needed to fully elucidate how other intratumoral therapies, including other TLR agonists, may impact T cell exhaustion and therapeutic efficacy in distant tumors. Models of metastatic seeding (e.g., to the liver and lung) would be of particular interest to address therapeutic efficacy in tissues with disparate immune microenvironments. Novel TLR9 agonists, for example, are thought to induce a systemic effect by activating plasmacytoid DCs (pDCs) (42–44). Whether the pDCs trigger an immune response through direct antigen presentation versus the secretion of type 1 IFN alone and how this may impact T cell exhaustion in distant tumors are questions of significant interest. On the basis of the results described here, we are currently preparing prospective clinical trials to test the CMP regimen in patients with various solid malignancies. Methods Cell lines and tumor challenge experiments. Female C57BL/6 WT and Rag1–/– mice aged 6 to 8 weeks were purchased from The Jackson Laboratory. Batf3–/– mice were a gift from L. Deng (MSKCC, New York, New York, USA), and Pmel-1 TCR–transgenic mice were a gift from N. Restifo (NCI, Bethesda, Maryland, USA). The cell lines B16F10, LLC, and Hep-55.1C were obtained from ATCC, and the MC38 line was obtained from the NCI. All cell lines were confirmed to be mycoplasma negative by the MSKCC Antibody and Bioresource Core facility. Cells were maintained in RPMI 1640 supplemented with 10% FCS, 1× nonessential amino acids, 1 mM sodium pyruvate, 2 mM l-glutamine, and penicillin with streptomycin (complete RPMI media). KPC tumor cells (1 × 106) were surgically implanted orthotopically into the tail of the pancreas 8 days prior to initial treatment, other tumors were implanted by intradermal injection of 5 × 105 cells into the flanks. Prior to treatment, mice were randomized and then treated twice weekly for 4 weeks. Tumors were measured twice weekly and monitored for at least 90 days after tumor challenge. In vivo reagent and treatments. Therapeutic in vivo mAbs anti–PD-1 (RMP1-14) and anti-CD40 (FGK45), corresponding IgG isotype controls (2A3), and depleting mAbs anti-CD4 (GK1.5) and anti-CD8+ (2.43) were purchased from Bio X Cell. RMP1-14 (250 μg) and 2A3 (250 μg) were administered intraperitoneally twice weekly. FGK45 (20 μg), MPL (5 μg), and 2A3 (20 μg) were administered concurrently intratumorally twice weekly. MPL (Sigma-Aldrich) was reconstituted as previously described (45). Depleting mAbs GK1.5 (560 μg) and 2.43 (400 μg) were administered intraperitoneally twice weekly beginning 1 day prior to treatment initiation and continued for the duration of the experiment. In vivo phagocytosis. B16F10 tumor–bearing mice were injected intratumorally with the treatments described, together with 2 × 107 FITC-conjugated 1-μm latex particles (Polysciences) as previously described (11). Twenty-four hours after treatment, DLNs were harvested and analyzed by flow cytometry. In vitro studies. For in vitro phagocytosis experiments, mice were sacrificed and bone marrow from tibiae and femurs was harvested and cultured in the presence of 20 ng/μL GM-CSF in T cell growth medium (complete RPMI media with 50 μM β-mercaptoethanol). On day 3, the supernatant was discarded and replaced with fresh media and GM-CSF. On day 6, the supernatant was discarded, and adherent cells were physically dissociated. The cells were then plated with FITC-conjugated 1-μm latex particles (Polysciences) and treated with either isotype, MPL (5 μg), or anti-CD40 (20 μg) for 24 hours before analysis by flow cytometry. For cell viability studies, when B16F10 cells reached 40% confluence, media were replaced with fresh media containing 20 μg/mL of either MPL, anti-CD40, or gemcitabine. After a 72-hour incubation, cells were assayed with the CellTiter-Glo Luminescent Cell Viability Assay (Promega) as previously described (46). Immune cell isolation and flow cytometry. Immune cells were isolated and subsequently analyzed by flow cytometry as previously described (47). Briefly, tumors and LNs were harvested by dissection from sacrificed mice and mechanically homogenized into single-cell suspensions filtered through 100-μm nylon filters (BD Biosciences) into cold RPMI supplemented with 7.5% FCS. Cell suspensions were then washed once with cold RPMI. Spleens were processed in the same way with additional RBC lysis using ACK Lysing Buffer (Lonza). All samples were resuspended in PBS with 0.5% (v/v) FCS. Samples were preincubated with anti-CD16/32 mAb (Fc block, clone 2.4G, BD Biosciences) for 15 minutes at 4°C to prevent nonspecific Fc receptor binding. For surface markers, samples were then stained for 30 minutes at 4°C with various combinations of fluorochrome-conjugated antibodies: anti–CD45–Pacific Orange (clone 30-F11, Invitrogen, Thermo Fisher Scientific); anti–CD8+-APC-Cy7 (clone 53-6.7, BD Biosciences); anti–CD4-AF700 (clone RM4-5, BD Biosciences); anti-CD11c-AF700 (clone N418, eBioscience); anti–CD86-PE (clone GL1, eBioscience); anti–PD-1–FITC (RMP1-30, eBioscience); anti–Tim-3–BV480 (clone 5D12, BD Biosciences); anti–Lag-3–APC (clone C9B7W, BioLegend); and anti–2B4-PerCP-Cy5.5 (clone 2B4, BioLegend). For intracellular stain, cells were permeabilized using a FoxP3 Fixation and Permeabilization Kit (eBioscience) and stained with various combinations of the following antibodies: anti–Ki67-PE/Dazzle 594 (clone 16A8, BioLegend); anti–Eomes-PE (clone Dan11mag, eBioscience); anti–IFN-γ-V450 (clone XMG1.2, BD Biosciences); and anti–granzyme B–PE-Cy7 (clone NGZB, eBioscience). All antibodies were purchased from BD, eBioscience, or Invitrogen (Thermo Fisher Scientific). For intracellular cytokine staining, mouse immune cells were restimulated with 500 ng/mL PMA and 1 μg/mL ionomycin in T cell growth medium (complete RPMI media with 50 μM β-mercaptoethanol) at 37°C. After 1 hour, 1× GolgiStop and 1× GolgiPlug (BD Biosciences) were added and incubated for an additional 4–6 hours at 37°C. Fc blockade, surface staining, and intracellular staining were then performed as described above. Stained cells were acquired using an LSR II Flow Cytometer or LSRFortessa X-50 and BD FACSDiva software (BD Biosciences). The data were further analyzed with FlowJo software (version 10.4). Debris and doublets were excluded on the basis of forward and side scatter measurements. Dead cells were excluded using Fixable Viability Dye eFluor 506 (eBioscience). Transcriptome profiling and gene set enrichment analysis. Bilateral B16F10 tumor–bearing B57BL/6 mice were treated as described above. Four animals per treatment group were sacrificed at 24 hours or 7 days. Bilateral tumors, DLNs, and spleens were harvested and snap-frozen in liquid nitrogen. Organs were then thawed, and total RNA was extracted with TRIzol (Invitrogen, Thermo Fisher Scientific). Expression profiling was performed using Affymetrix Clariom S and analyzed using the Transcriptome Analysis Console as previously described (48, 49). All original transcriptomics data were deposited in the NCBI’s Gene Expression Omnibus (GEO) database (GEO GSE130027). Gene set enrichment was performed using GAGE (48, 49) on the robust multichip average normalized transcript expression values to identify significant gene set enrichment from immunologic signatures in the Molecular Signatures Database (48, 49) at a FDR of less than 0.05. IFN-γ ELISPOT assay. Splenocytes were harvested from B16F10 tumor–bearing mice 1 week after initial treatment, and a single-cell suspension was generated as described above. CD8+ T cells were isolated using a CD8+ T cell Isolation Kit (Miltenyi Biotec) according to the manufacturer’s instructions. IFN-γ–producing T cells were quantified as previously described (47). Briefly, the Mouse ImmunoSpot IFN-γ Single Color ELISPOT system was used (Cellular Technology Limited). For in vitro restimulation, 1 × 105 CD8+ T cells were cocultured with 1 × 105 irradiated (60 Gy) B16F10 target cells for 16 hours. Irradiated syngeneic ID8 ovarian carcinoma target cells were used as negative control targets to assess specificity. IFN-γ spots were quantified using a ImmunoSpot S6 Micro Analyzer and ImmunoSpot Professional Software (both from Cellular Technology Limited). Killing assays. For clonogenic ex vivo killing assays (24), a 48-well tissue culture plate was filled sequentially with 5 μL PBS containing 0.1 U thrombin, 1 mg/mL human fibrinogen in 100 μL PBS, 1 mg/mL rat tail collagen I, 10% FCS, and B16F10 cells, with or without magnetic-activated cell-sorting–purified (MACS-purified) CD8+ T cells at a 100:1 ratio for splenic CD8+ T cells and 10:1 for tumor CD8+ T cells. The plates were incubated for 20 minutes at 37°C in a 95% air and 5% CO2 humidified atmosphere to allow the fibrin to gel. Gels were overlaid with 1 mL complete RPMI media with 50 μM β-mercaptoethanol and incubated at 37°C in a 95% air and 5% CO2 humidified atmosphere. Twenty-four hours later, the gels were lysed by sequential collagenase (2.5 mg/mL) and trypsin (2.5 mg/mL; Sigma-Aldrich) digestion. The lysed gels were then diluted, and the recovered melanoma cells were plated in 6-well plates for colony formation. After 7 days in culture, the plates were fixed with formaldehyde and stained with 2% methylene blue, and the colonies were counted manually. Target cell killing was calculated using the equation: 1 − (melanoma + T cells)/(melanoma alone). The killing coefficient k was calculated by applying the following equation: b = b0 e − (kp − g)t in which b is the tumor concentration at any time, b0 is the initial tumor concentration, p is the T cell concentration, k is the second-order rate constant for T cell killing of tumor, and g is the first-order rate constant for tumor growth. Adoptive transfer. CD8+ T cells were purified from spleens and LNs from Pmel-1 TCR–transgenic mice (23) using magnetic beads (Miltenyi Biotec) for positive selection as described above. Transgenic CD8+ T cells were then loaded with CellTrace Violet (CTV) (Thermo Fisher Scientific) according to the manufacturer’s instructions. On day 7 after tumor challenge, 2 × 106 CTV-labeled Pmel-1 CD8+ T cells were administered by tail-vein injection into the mice. A single treatment with CMP or isotype control was administered on day 8, and LNs were harvested on day 11. Proliferation was then assessed by flow cytometry. Multidimensional encoding of MHC multimers. Murine gp100 peptide (EGSRNQDWL, AnaSpec) was exchanged onto H2-Db monomers (courtesy of the NIH Tetramer Facility at Emory University, Atlanta, Georgia, USA) complexed with UV-cleavable peptides as previously described (50). Briefly, excess gp100 peptide was plated with UV-cleavable monomers. The monomers were destabilized by the cleavage but rescued by the presence of gp100 peptide. Monomers bearing gp100 were then conjugated via streptavidin-biotin interaction to 1 of 2 distinct fluorophores (BV786 and BB515, BD Biosciences). Distant tumors were harvested from tumor-bearing mice after 1 week of treatment. Samples were stained with 1 μg/mL multimer at 37°C for 15 minutes. Cells were rinsed with PBS with 0.5% (v/v) FCS, followed by surface staining with and 100 ng/mL BV805-labeled anti-CD8+ mAbs (BD Bioscience) for 30 minutes at 4°C. Analysis by flow cytometry on an LSR II (BD Bioscience) was performed, and the data obtained were analyzed using FlowJo software (version 10.4). Live CD8+ T cells that stained positive for both fluorophores were considered specific for gp100. . Differential gene expression and survival analyses. To correlate transcriptomic changes derived from preclinical models to patient samples, we first conducted differential gene expression analysis of our microarray data between replicates in the distant tumor at 1 week with CMP versus isotype (n = 4) using limma (version 3.38.2) (51). Genes differentially expressed at a FDR P value of 0.1 and a log fold-change of more than 0 (i.e., increased in CMP) were converted to human Entrez IDs using clusterProfiler (version 3.8.1) (52). This yielded a final gene set of 73 genes (Supplemental Table 1) for evaluation of patients with pre-therapy RNA-Seq data from the Riaz et al. anti–PD-1 cohort (n = 51) (33). Fragments per kilobase per million mapped reads (FPKMs) for patients in the Riaz et al. cohort were calculated using the “fpkm()” function in DESeq2 (version 1.22.1) (53) and publicly available scripts in GitHub (https://github.com/riazn/bms038_analysis; commit ID: 137111c). To create a binary variable for survival analysis, we calculated the mean FPKM across all genes in the gene set and stratified patients into high and low expression groups using the top quartile of the distribution of mean gene set FPKMs as a cutoff. Progression-free survival, HRs, and CIs were calculated using Survival (version 2.42.6), and statistical significance was calculated using the log-rank test. All analyses were conducted in R (version 3.5.0) (https://www.R-project.org/). Statistics. Data were analyzed for statistical significance with an unpaired, 2-tailed Student’s t test when comparing the means of 2 independent groups. All data represent the mean ± SEM. A P value of less than 0.05 was considered statistically significant. In experiments with multiple t test correction, P values were adjusted using the Holm-Sidak method. Progression-free survival of nivolumab-treated patients described here (33) was estimated using the Kaplan-Meier method. All survival data were analyzed by log-rank (Mantel-Cox) test. All experiments were repeated 2–4 times. Study approval. All mouse procedures were conducted in accordance with protocols and guidelines established at the MSKCC, and all mouse procedures and experiments were approved by the IACUC of MSKCC. Mice were maintained according to NIH animal care guidelines, under a protocol approved by the IACUC of the MSKCC. Author contributions DNK and NS designed and performed experiments, analyzed the data, and prepared the manuscript. LFC, SB, DH, RZ, RG, BG, MS, AG, and JAM designed and performed experiments as well as analyzed data. DR, RMS, CK, KSP, MC, and SH conducted bioinformatics and statistical analyses. JJH and VPB assisted with manuscript preparation. DNK, JDW, and TM oversaw the experimental design, data interpretation, and manuscript preparation. Supplemental material View Supplemental data Acknowledgments We thank the NIH Tetramer Facility for providing multimers used in this study. This research was funded in part through the NIH, NCI Cancer Center Support Grant P30 CA008748; NCI R01 CA056821; the Ludwig Collaborative and Swim Across America Laboratory; the Emerald Foundation; the Tri-Institutional Therapeutics Discovery Institute; the Memorial Sloan Kettering Technology Development Fund, MSKCC; the Parker Institute for Cancer Immunotherapy, MSKCC; the Department of Medicine, MSKCC; and Weill Cornell Medicine. RG acknowledges support from an NIH-T32 Postdoctoral Research Fellowship. Footnotes Conflict of interest: DNK, TM, and JDW are co-inventors on patent applications related to CD40 and in situ vaccination (PCT/US2016/045970), filed by MSKCC. JDW is a paid consultant for Advaxis, Bristol-Myers Squibb, Merck, Medimmune, Celgene, and Genentech and receives research funding from Bristol-Myers Squibb, Merck, Genentech, and Medimmune and honoraria from Ono Pharmaceutical Company. JDW is a cofounder of, paid consultant for, and stock option owner in Potenza Therapeutics, Tizona Therapeutics, and IMVAQ Therapeutics. JDW is a paid consultant for and has stock option ownership in BeiGene and Apricity. JDW is a paid consultant for Surface Oncology, Polaris, Polynoma, Array, Ascentage Pharma, PureTech, Chugai Pharmaceutical, FStar, Amgen, SELLAS Life Sciences, Serametrix, Neon, Eli Lilly, PsiOxus Therapeutics, Syndax Pharmaceuticals, Recepta, Amgen, and Puretech and reports stock option ownership in Adaptive Biotechnologies. JDW is an inventor on the following patents: xenogeneic DNA vaccines; alphavirus replicon particles expressing TRPTM is a consultant for Immunos Therapeutics and Pfizer. TM is a cofounder of and equity holder in IMVAQ Therapeutics. TM receives research funding from Bristol-Myers Squibb, Surface Oncology, Kyn Therapeutics, Infinity Pharmaceuticals, Peregrine Pharmaceuticals, Adaptive Biotechnologies, Leap Therapeutics, and Aprea Therapeutics. TM is an inventor on patent applications related to work on oncolytic viral therapy; alpha virus–based vaccine; neo antigen modeling; CD40, GITR, OX40, PD-1, and CTLA-4. LFC is a consultant for Merck. SB has received royalties from Agenus. RZ is an inventor on patent applications related to work on GITR, PD-1, and CTLA-JJH is a consultant for Bristol-Myers Squib, Eisai, Elly Lilly, and CytomX Therapeutics and has received research support from Bristol-Myers Squib. VPB is a recipient of an immune-oncology translational research grant from Bristol-Myers Squibb and is an inventor on a patent application related to work on neoantigen modeling. Copyright: © 2019, American Society for Clinical Investigation. Reference information: J Clin Invest. https://doi.org/10.1172/JCI128562. References Rizvi H, et al. Molecular determinants of response to anti-programmed cell death (PD)-1 and anti-programmed death-ligand 1 (PD-L1) blockade in patients with non-small-cell lung cancer profiled with targeted next-generation sequencing. J Clin Oncol. 2018;36(7):633–641. View this article via: PubMed CrossRef Google Scholar Tumeh PC, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515(7528):568–571. View this article via: PubMed CrossRef Google Scholar Pauken KE, Wherry EJ. Overcoming T cell exhaustion in infection and cancer. Trends Immunol. 2015;36(4):265–276. View this article via: PubMed CrossRef Google Scholar Hui E, et al. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science. 2017;355(6332):1428–1433. View this article via: PubMed CrossRef Google Scholar Kamphorst AO, et al. Rescue of exhausted CD8 T cells by PD-1-targeted therapies is CD28-dependent. Science. 2017;355(6332):1423–1427. View this article via: PubMed CrossRef Google Scholar Lanier LL, et al. CD80 (B7) and CD86 (B70) provide similar costimulatory signals for T cell proliferation, cytokine production, and generation of CTL. J Immunol. 1995;154(1):97–105. View this article via: PubMed Google Scholar Fend L, et al. Immune checkpoint blockade, immunogenic chemotherapy or IFN-α blockade boost the local and abscopal effects of oncolytic virotherapy. Cancer Res. 2017;77(15):4146–4157. View this article via: PubMed CrossRef Google Scholar Ager CR, Reilley MJ, Nicholas C, Bartkowiak T, Jaiswal AR, Curran MA. Intratumoral STING activation with T cell checkpoint modulation generates systemic antitumor immunity. Cancer Immunol Res. 2017;5(8):676–684. View this article via: PubMed CrossRef Google Scholar Mata-Haro V, Cekic C, Martin M, Chilton PM, Casella CR, Mitchell TC. The vaccine adjuvant monophosphoryl lipid A as a TRIF-biased agonist of TLR4. Science. 2007;316(5831):1628–1632. View this article via: PubMed CrossRef Google Scholar Vernacchio L, et al. Effect of monophosphoryl lipid A (MPL) on T-helper cells when administered as an adjuvant with pneumocococcal-CRM197 conjugate vaccine in healthy toddlers. Vaccine. 2002;20(31-32):3658–3667. View this article via: PubMed CrossRef Google Scholar Piconese S, Valzasina B, Colombo MP. OX40 triggering blocks suppression by regulatory T cells and facilitates tumor rejection. J Exp Med. 2008;205(4):825–839. View this article via: PubMed CrossRef Google Scholar Caux C, et al. Activation of human dendritic cells through CD40 cross-linking. J Exp Med. 1994;180(4):1263–1272. View this article via: PubMed CrossRef Google Scholar Guiducci C, Vicari AP, Sangaletti S, Trinchieri G, Colombo MP. Redirecting in vivo elicited tumor infiltrating macrophages and dendritic cells towards tumor rejection. Cancer Res. 2005;65(8):3437–3446. View this article via: PubMed CrossRef Google Scholar Andtbacka RH, et al. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J Clin Oncol. 2015;33(25):2780–2788. View this article via: PubMed CrossRef Google Scholar Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci U S A. 2010;107(9):4275–4280. View this article via: PubMed CrossRef Google Scholar Fu J, Malm IJ, Kadayakkara DK, Levitsky H, Pardoll D, Kim YJ. Preclinical evidence that PD1 blockade cooperates with cancer vaccine TEGVAX to elicit regression of established tumors. Cancer Res. 2014;74(15):4042–4052. View this article via: PubMed CrossRef Google Scholar Lollini PL, et al. Enhancement of experimental metastatic ability by tumor necrosis factor-alpha alone or in combination with interferon-gamma. Clin Exp Metastasis. 1990;8(2):215–224. View this article via: PubMed CrossRef Google Scholar Engelhardt JJ, et al. Marginating dendritic cells of the tumor microenvironment cross-present tumor antigens and stably engage tumor-specific T cells. Cancer Cell. 2012;21(3):402–417. View this article via: PubMed CrossRef Google Scholar Randolph GJ, Angeli V, Swartz MA. Dendritic-cell trafficking to lymph nodes through lymphatic vessels. Nat Rev Immunol. 2005;5(8):617–628. View this article via: PubMed CrossRef Google Scholar Hildner K, et al. Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science. 2008;322(5904):1097–1100. View this article via: PubMed CrossRef Google Scholar Fuertes MB, et al. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells. J Exp Med. 2011;208(10):2005–2016. View this article via: PubMed CrossRef Google Scholar Murphy TL, Tussiwand R, Murphy KM. Specificity through cooperation: BATF-IRF interactions control immune-regulatory networks. Nat Rev Immunol. 2013;13(7):499–509. View this article via: PubMed CrossRef Google Scholar Overwijk WW, et al. Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. J Exp Med. 2003;198(4):569–580. View this article via: PubMed CrossRef Google Scholar Budhu S, et al. CD8+ T cell concentration determines their efficiency in killing cognate antigen-expressing syngeneic mammalian cells in vitro and in mouse tissues. J Exp Med. 2010;207(1):223–235. View this article via: PubMed CrossRef Google Scholar Andersen RS, et al. Parallel detection of antigen-specific T cell responses by combinatorial encoding of MHC multimers. Nat Protoc. 2012;7(5):891–902. View this article via: PubMed CrossRef Google Scholar Byrne KT, Vonderheide RH. CD40 stimulation obviates innate sensors and drives T cell immunity in cancer. Cell Rep. 2016;15(12):2719–2732. View this article via: PubMed CrossRef Google Scholar Joeckel LT, et al. Mouse granzyme K has pro-inflammatory potential. Cell Death Differ. 2011;18(7):1112–1119. View this article via: PubMed CrossRef Google Scholar Wensink AC, et al. Granzyme K synergistically potentiates LPS-induced cytokine responses in human monocytes. Proc Natl Acad Sci U S A. 2014;111(16):5974–5979. View this article via: PubMed CrossRef Google Scholar Doering TA, Crawford A, Angelosanto JM, Paley MA, Ziegler CG, Wherry EJ. Network analysis reveals centrally connected genes and pathways involved in CD8+ T cell exhaustion versus memory. Immunity. 2012;37(6):1130–1144. View this article via: PubMed CrossRef Google Scholar Barber DL, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439(7077):682–687. View this article via: PubMed CrossRef Google Scholar Paley MA, et al. Progenitor and terminal subsets of CD8+ T cells cooperate to contain chronic viral infection. Science. 2012;338(6111):1220–1225. View this article via: PubMed CrossRef Google Scholar Totsuka T, et al. Regulation of murine chronic colitis by CD4+CD25- programmed death-1+ T cells. Eur J Immunol. 2005;35(6):1773–1785. View this article via: PubMed CrossRef Google Scholar Riaz N, et al. Tumor and microenvironment evolution during immunotherapy with nivolumab. Cell. 2017;171(4):934–949.e16. View this article via: PubMed CrossRef Google Scholar Postow MA, et al. Immunologic correlates of the abscopal effect in a patient with melanoma. N Engl J Med. 2012;366(10):925–931. View this article via: PubMed CrossRef Google Scholar Zamarin D, et al. Intratumoral modulation of the inducible co-stimulator ICOS by recombinant oncolytic virus promotes systemic anti-tumour immunity. Nat Commun. 2017;8:14340. View this article via: PubMed Google Scholar Zamarin D, et al. Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci Transl Med. 2014;6(226):226ra32. View this article via: PubMed CrossRef Google Scholar Dai P, et al. Intratumoral delivery of inactivated modified vaccinia virus Ankara (iMVA) induces systemic antitumor immunity via STING and Batf3-dependent dendritic cells. Sci Immunol. 2017;2(11):eaal1713. View this article via: PubMed Google Scholar Marabelle A, Tselikas L, de Baere T, Houot R. Intratumoral immunotherapy: using the tumor as the remedy. Ann Oncol. 2017;28(suppl_12):xii33–xii43. View this article via: PubMed CrossRef Google Scholar Ulrich JT, Myers KR. Monophosphoryl lipid A as an adjuvant. Past experiences and new directions. Pharm Biotechnol. 1995;6:495–524. View this article via: PubMed CrossRef Google Scholar Beatty GL, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. 2011;331(6024):1612–1616. View this article via: PubMed CrossRef Google Scholar Vidal L, et al. A phase I study of intravenous oncolytic reovirus type 3 Dearing in patients with advanced cancer. Clin Cancer Res. 2008;14(21):7127–7137. View this article via: PubMed CrossRef Google Scholar Krieg AM. Toll-like receptor 9 (TLR9) agonists in the treatment of cancer. Oncogene. 2008;27(2):161–167. View this article via: PubMed CrossRef Google Scholar Parker BS, Rautela J, Hertzog PJ. Antitumour actions of interferons: implications for cancer therapy. Nat Rev Cancer. 2016;16(3):131–144. View this article via: PubMed CrossRef Google Scholar Ribas A, et al. SD-101 in combination with pembrolizumab in advanced melanoma: results of a phase Ib, multicenter study. Cancer Discov. 2018;8(10):1250–1257. View this article via: PubMed CrossRef Google Scholar Stark R, Choi H, Koch S, Lamb F, Sherwood E. Monophosphoryl lipid A inhibits the cytokine response of endothelial cells challenged with LPS. Innate Immun. 2015;21(6):565–574. View this article via: PubMed CrossRef Google Scholar Thoreen CC, et al. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J Biol Chem. 2009;284(12):8023–8032. View this article via: PubMed CrossRef Google Scholar De Henau O, et al. Overcoming resistance to checkpoint blockade therapy by targeting PI3Kγ in myeloid cells. Nature. 2016;539(7629):443–447. View this article via: PubMed CrossRef Google Scholar Nassal DM, et al. KChIP2 is a core transcriptional regulator of cardiac excitability. Elife. 2017;:e17304. View this article via: PubMed Google Scholar Andrzejewski S, et al. PGC-1α promotes breast cancer metastasis and confers bioenergetic flexibility against metabolic drugs. Cell Metab. 2017;26(5):778–787.e5. View this article via: PubMed CrossRef Google Scholar Rodenko B, et al. Generation of peptide-MHC class I complexes through UV-mediated ligand exchange. Nat Protoc. 2006;1(3):1120–1132. View this article via: PubMed CrossRef Google Scholar Ritchie ME, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47. View this article via: PubMed CrossRef Google Scholar Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16(5):284–287. View this article via: PubMed CrossRef Google Scholar Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. View this article via: PubMed CrossRef Google Scholar Version history Version 1 (July 22, 2019): Electronic publication
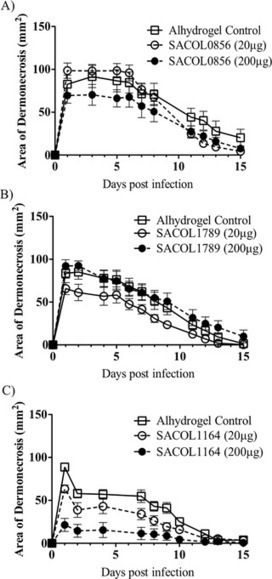
Staphylococcus aureus infections represent a major public health threat, but previous attempts at developing a universal vaccine have been unsuccessful. We attempted to identify a vaccine that would be protective against both skin/soft tissue and bloodstream infections. We first tested a panel of staphylococcal antigens that are conserved across strains, combined with aluminum hydroxide as an adjuvant, for their ability to induce protective immunity in both skin and bacteremia infection models. Antigens were identified that reduced dermonecrosis during skin infection, and other non-overlapping antigens were identified that showed trends to protection in the bacteremia model. However, individual antigens were not identified that mediated substantial protection in both the skin and bacteremia infection models. We therefore tested a variety of combinations of proteins to seek a single combination that could mediate protection in both models. After iterative testing, a vaccine consisting of 3 antigens, ABC transporter protein (SACOL2451), ABC2 transporter protein (SACOL0695), and α-hemolysin (SACOL1173), was identified as the most effective combination. This combination vaccine provided protection in a skin infection model. However, these antigens were only partially protective in the bacteremia infection model. Even by testing multiple different adjuvants, optimized efficacy in the skin infection model did not translate into efficacy in the bacteremia model. Thus protective vaccines against skin/soft tissue infections may not enable effective protection against bloodstream infections.
|


















recent snippets of information on https://www.scoop.it/topic/immunology-and-biotherapies?q=vaccine