
ADC is a promising cancer therapeutic...
Get Started for FREE
Sign up with Facebook Sign up with X
I don't have a Facebook or a X account


 Your new post is loading...
Your new post is loading... Your new post is loading...
Your new post is loading...
Research ArticleOncologyTherapeutics Free access | 10.1172/jci.insight.123281 Tofacitinib enhances delivery of antibody-based therapeutics to tumor cells through modulation of inflammatory cells Nathan Simon,1 Antonella Antignani,1 Stephen M. Hewitt,1 Massimo Gadina,2 Christine Alewine,1 and David FitzGerald1 First published February 5, 2019 - More info Abstract The routes by which antibody-based therapeutics reach malignant cells are poorly defined. Tofacitinib, an FDA-approved JAK inhibitor, reduced tumor-associated inflammatory cells and allowed increased delivery of antibody-based agents to malignant cells. Alone, tofacitinib exhibited no antitumor activity, but combinations with immunotoxins or an antibody-drug conjugate resulted in increased antitumor responses. Quantification using flow cytometry revealed that antibody-based agents accumulated in malignant cells at higher percentages following tofacitinib treatment. Profiling of tofacitinib-treated tumor-bearing mice indicated that cytokine transcripts and various proteins involved in chemotaxis were reduced compared with vehicle-treated mice. Histological analysis revealed significant changes to the composition of the tumor microenvironment, with reductions in monocytes, macrophages, and neutrophils. Tumor-associated inflammatory cells contributed to non-target uptake of antibody-based therapeutics, with mice treated with tofacitinib showing decreased accumulation of therapeutics in intratumoral inflammatory cells and increased delivery to malignant cells. The present findings serve as a rationale for conducting trials where short-term treatments with tofacitinib could be administered in combination with antibody-based therapies. Introduction Overcoming treatment resistance is a major goal of cancer research. Various cell types within the tumor microenvironment (TME) have been posited as barriers to effective treatment including macrophages (1, 2), monocytes (3), fibroblasts (4), neutrophils (5), regulatory T cells (6), and endothelial cells (7). These stromal cells, together with secreted factors, can protect malignant cells from a variety of therapies including cytotoxic drugs (7, 8). Likewise, the adaptive immune system works poorly in an immunosuppressive tumor environment where alternatively polarized macrophages, myeloid-derived suppressor cells (MDSCs), or regulatory T cells are present (9–11). Aberrant vascular architecture leading to hypoxia and irregular vessel formation, resulting in poor drug delivery, has prompted the development of vasculature-normalizing agents with the goal of improving access to malignant cells (7, 12). When antibody-based agents are employed, high levels of shed antigen or antigen-negative variants can result in poor treatment outcomes (13, 14). In addition, there is speculation that high numbers of inflammatory cells can interfere with delivery to malignant cells (15). To address this, here we investigate antibody delivery to malignant cells following treatment of tumor-bearing mice with the JAK/STAT inhibitor, tofacitinib. So-called naked antibodies, including rituximab, cetuximab, and trastuzumab, were some of the first antibody-based therapeutics to be developed and have been approved for many years. More recently, antibody-drug conjugates (ADCs), immunotoxins, chimeric antigen receptor (CAR) T cells, bispecific T cell engagers (BiTEs), and checkpoint inhibitor antibodies have been approved. With the exception of checkpoint inhibitors, antibody therapeutics must be delivered directly to malignant cells where they bind surface antigens or receptors. Antibodies are effective either because they nullify essential growth signals, recruit effector cells, or deliver cytotoxic payloads. However, failure to contact malignant cells is likely to lead to a poor response. The routes by which antibodies reach malignant cells within the larger TME have not been extensively documented. Desmoplastic tumors like pancreatic or triple-negative breast cancers (TNBCs) are characterized by abundant extracellular matrix (ECM) and tumor infiltration of inflammatory immune cell populations like MDSCs and tumor-associated macrophages or neutrophils (TAMs or TANs), which can hinder therapeutic efficacy (16, 17). In addition to aggressive growth and metastatic potential, pancreatic ductal adenocarcinomas (PDACs) and TNBCs lack effective targeted therapies, instead relying on conventional chemotherapies that often result in drug-resistant relapse and progression (18, 19). PDACs have a 5-year survival rate of 5%–10% (20) and disease severity correlates with increased inflammation and stromal contributions to tumor growth (21, 22). In particular, the presence of TANs is correlated with poorer outcomes and resistance to chemotherapies and checkpoint inhibitors (23). Like pancreatic cancers, TNBC progression relies on significant contribution from the tumor stroma and inflammatory cell populations, with accumulation of collagen-rich fibrotic lesions and the subsequent release of neutrophil chemoattractants from fibroblasts responsible for recruiting inflammatory cells (24, 25). However, monocytes and neutrophils are relatively short-lived, requiring continuous release of cytokines from the growing tumor to recruit and replenish these cells. If cytokines could be downregulated, the TME would undoubtedly be altered, possibly allowing a more favorable situation for therapeutic intervention, including improved delivery of antibody-based agents. Integral to the development of antibody-based agents is the necessity to identify surface antigens expressed prominently or exclusively on tumorigenic cells, but not on vital normal tissues (26–28). These antigens have become targets for such antibody-directed therapeutics as immunotoxins and ADCs (29). Immunotoxins consist of an antibody fragment or receptor ligand targeting a surface antigen/receptor joined to a plant or bacterial toxin component, typically ricin or saporin, diphtheria toxin, or Pseudomonas aeruginosa exotoxin A (PE) (30). The catalytic inhibition of protein synthesis makes immunotoxins highly potent, with estimates suggesting that a only small number of toxin molecules delivered to the cytosol is sufficient to kill a susceptible cell (31). This potency has led to the testing of immunotoxins in clinical trials for treatment of aggressive tumors, including brain (32), mesothelioma (33), and pancreatic (34) cancers, as well as hematologic cancers (35–37). Specifically, the immunotoxin LMB-100, which targets surface mesothelin, is being evaluated in clinical trials for malignant mesothelioma and pancreatic cancer (NCT02810418 and NCT02798536). In contrast, ADCs are typically full-length antibodies conjugated to potent cytotoxic drugs that must be delivered in sufficient quantities to inhibit DNA replication or cell division (38). ADCs have also been tested in a variety of clinical settings, exhibiting several notable successes (39, 40). However, all antibody-based therapeutics rely on efficient delivery to the malignant cell population, as inefficient delivery often necessitates increased dosing, risking significant off-target toxicities. With this as background, we set out to evaluate the in vivo activity of the pan-JAK inhibitor tofacitinib, in combination with antibody-based therapeutics targeting pancreatic cancer and TNBC models. The initial rationale for this evaluation came from earlier studies showing that tofacitinib could reduce antidrug immune responses that often accompany the administration of immunotoxins to individuals or test animals that have an intact immune system (41). In addition, rheumatoid arthritis (RA) patients treated with tofacitinib show potent inhibition of JAK/STAT signaling and a subsequent decrease in circulating proinflammatory cytokines resulting in fewer inflammatory cells within arthritic lesions (42, 43). As tofacitinib inhibits both proinflammatory signaling and the antidrug immune response, it was evaluated in combination with immunotoxin therapy. These experiments were then extended to include a mesothelin-targeted ADC currently under clinical development (44). We report that tofacitinib enhanced the antitumor activity of both immunotoxins and the ADC. Mechanistic insights suggested that enhancement was associated with decreased cytokine production and decreased recruitment of TAMs and TANs to the tumor. In turn, this resulted in a decrease in off-target uptake of therapeutic molecules by these inflammatory cell populations, leading to enhanced delivery of antibody-based agents to the malignant cells. These preclinical results provide a rationale for human trials combining tofacitinib with a variety of antibody-based therapeutics. Results Tofacitinib enhances immunotoxin activity in vivo. In humans, the pan-JAK inhibitor tofacitinib reduces joint inflammation and immune cell infiltrates associated with RA (42, 43, 45). In mice, tofacitinib reduces antibody responses to foreign proteins including immunotoxins (41); therefore, it was of interest to test tofacitinib in combination with the antitumor activity of immunotoxins, first in a xenograft model of TNBC. MDA-MB-468 tumors were treated with vehicle, tofacitinib alone, immunotoxin alone, or a combination of both. Tumors in vehicle- or tofacitinib-treated groups (n = 6 per group) quickly reached the experimental endpoint of 1,200 mm3 (Figure 1A), with median survival of 33.5 and 36.5 days, respectively (P = 0.478) (Figure 1B). Tumors in mice treated with the immunotoxin (HB21-PE40, targeting the human transferrin receptor) displayed a brief but significant delay in tumor growth (Figure 1A). Median survival for immunotoxin-treated mice was 47 days, significantly improved over vehicle-treated mice (P < 0.001) (Figure 1B). Treatment of tumors with the combination therapy resulted in enhanced antitumor activity, with an initial decrease in tumor volume during treatment followed by a sustained delay of tumor growth for several weeks (P < 0.001) (Figure 1A). Median survival of combination-treated mice was significantly longer than mice treated with immunotoxin alone, with a median time to death of 64 days (P < 0.001) (Figure 1B). A second experimental replicate showed similar results (Supplemental Figure 1, A and C; supplemental material available online with this article; https://doi.org/10.1172/jci.insight.123281DS1). Figure 1 Tofacitinib enhances immunotoxin-mediated antitumor activity. (A) Mice bearing MDA-MB-468 TNBC xenografts were treated with vehicle, tofacitinib alone, immunotoxin (HB21-PE40) alone, or a combination of both treatments (n = 6–7 mice per treatment group). Mice received a single treatment cycle (once every other day; arrows). Significance was calculated by 2-tailed t test at experimental endpoints. Data are representative of 2 independent experiments, showing similar results. (B) Kaplan-Meyer plot displaying time to endpoint for each treatment group of mice implanted with MDA-MB-468 tumors. Log-rank test was performed to calculate significance. (C) Mice bearing KLM-1 PDAC xenografts were treated with vehicle, tofacitinib alone, immunotoxin (LMB-100) alone, or a combination of both (n = 7–8 mice per treatment group). Mice received 2 cycles of treatment with a week in between cycles (each cycle: once per day, 4 times; arrows). Significance was calculated as above. (D) Kaplan-Meyer plot displaying time to endpoint of each treatment group for mice implanted with KLM-1 tumors. Log-rank test was performed to calculate significance. ***P < 0.001 compared with vehicle-treated; †P < 0.01 compared with immunotoxin treated (A–D). ns, not significant. Using the same treatment strategy, we next challenged the KLM-1 PDAC tumor model with the investigational immunotoxin LMB-100, which targets surface mesothelin (46). A preliminary experiment using only 1 treatment cycle proved less effective than in the TNBC model (Supplemental Figure 1, B and D). Therefore, we designed a second experiment whereby mice (n = 7 or 8 per group) received 2 cycles of treatment spaced 1 week apart (Figure 1C). As before, tumors in mice treated with vehicle or tofacitinib grew rapidly, with median time to endpoint of 28 days for each group (Figure 1D). Tumors treated with the LMB-100 immunotoxin showed reduced tumor growth compared with vehicle-treated tumors (P < 0.001), but no decrease in tumor volume and rapid regrowth after treatment. In contrast, tumors treated with LMB-100 plus tofacitinib showed a decrease in tumor volume during each treatment cycle, and delayed tumor regrowth after treatment. Median endpoint was not reached until 48 days, significantly extended from both vehicle- and tofacitinib-treated mice (P < 0.001) and from LMB-100–treated mice (P = 0.0013). Two combination-treated mice showed minimal tumor growth, but no complete regression and were sacrificed at 60 days. In both xenograft models, combination treatments were clearly more effective at inhibiting tumor growth than single agents. Tofacitinib does not enhance immunotoxin activity in vitro. To gain insight into the mechanism of increased immunotoxin activity, we investigated whether tofacitinib caused similar enhancements in tissue culture. Tofacitinib at concentrations of 1, 2, and 4 μM did not increase the cytotoxic activity of either immunotoxin targeting MDA-MB-468 or KLM-1 cells (Supplemental Figure 2, A and B). Tofacitinib alone, at concentrations up to 16 μM, had no appreciable effect on the viability of either cell line (Supplemental Figure 2, C and D) and also failed to enhance immunotoxin action (Supplemental Figure 2, E and F). Tofacitinib targets both JAK3 and JAK1 signaling and, to a lesser extent, JAK2 (47). However, cells of epithelial origin typically do not express JAK3 but can express either JAK1 or JAK2, which in turn can activate downstream STAT proteins. To determine if the lack of immunotoxin enhancement in tissue culture was due to poor JAK inhibition, we examined the status of downstream STAT proteins. Untreated, KLM-1 cells showed activation of both STAT1 and STAT3, whereas tofacitinib treatment eliminated detectable p-STAT3 (Supplemental Figure 2G). STAT1 activation was impacted only moderately. Similar results were obtained for MDA-MB-468 cells, which showed a robust p-STAT3 signal, consistent with the constitutive activation of STAT3 in many breast cancers (48) (Supplemental Figure 2H). Tofacitinib treatment completely abrogated p-STAT3, but did not alter STAT1 phosphorylation. Our results confirmed that tofacitinib inhibited the phosphorylation of relevant downstream JAK effectors in both cell lines and that failure to enhance immunotoxin action in tissue culture was unrelated to drug resistance or aberrant signaling pathways. Tofacitinib treatment enhances tumor uptake of antibody-based agents. As tofacitinib enhanced the antitumor activity of immunotoxins in vivo, but displayed no single-agent activity and no enhancement in vitro, we considered that tofacitinib was altering the TME, either permitting increased immunotoxin access to tumor cells or altering survival or proliferation signals. To examine the first possibility, we quantified the delivery of fluorescently labeled immunotoxin to tumor cells in xenograft models. Tumor-bearing mice were pretreated for approximately 70 hours with either vehicle or tofacitinib and then administered fluorescently labeled immunotoxin. Based on immunotoxin half-life and prior studies examining tumor uptake (49), tumors were excised 3 hours after injection, enzymatically dissociated into single-cell suspensions, and analyzed for immunotoxin uptake by flow cytometry (Supplemental Figure 3, A and B). Tumor cells were identified via gating with a human-specific antibody against the epidermal growth factor receptor (EGFR) to ensure that only uptake into malignant cells was analyzed. When KLM-1 tumors were retrieved from mice injected with fluorescently labeled LMB-100 (LMB-100–Alexa647), the average (n = 4 independent experiments) labeling of recovered malignant cells approached a median of 29% at 3 hours after injection. However, when mice were pretreated with tofacitinib, 43% of malignant cells exhibited an immunotoxin signal (Figure 2A) (P = 0.04). Similarly, mice bearing MDA-MB-468 tumors and treated with Alexa647-labeled HB21-PE40 for 3 hours showed increased tumor cell uptake of labeled immunotoxin in mice pretreated with tofacitinib compared with mice treated with vehicle (Figure 2B) (P = 0.05). These results confirmed a tofacitinib-mediated increase in the delivery of immunotoxin to tumor cells and suggested a potential explanation for the enhancement of immunotoxin activity. Figure 2 Tofacitinib enhances delivery of antibody-based therapeutics to cancer cells. (A) Mice bearing KLM-1 xenografts were injected with LMB-100–A647 for 3 hours with or without tofacitinib pretreatment. Human tumor cells were identified by incubation with anti-huEGFR–BV421. The average percentage of EGFR+ immunotoxin-labeled cells for each treatment is displayed. n = 4 independent biological replicates. (B) Mice bearing MDA-MB-468 xenografts were injected with HB21-PE40–Alexa647 for 3 hours with or without tofacitinib pretreatment. The percentage of EGFR+ immunotoxin-positive cells for each treatment is displayed. n = 4 independent biological replicates. (C) Mice with KLM-1 xenografts were injected with anti-EGFR–BV421 for 3 hours with or without tofacitinib pretreatment. Human tumor cells were identified by incubation with anti–human transferrin receptor–APC. The percentage of TfnR+ cells displaying EGFR antibody signal is displayed for each treatment. n = 2 independent biological replicates. (D) Mice with MDA-MB-468 xenografts were injected with anti-EGFR–BV421 for 3 hours with or without tofacitinib. The percentage of TfnR+ cells showing EGFR antibody signal is displayed for each treatment. n = 3 independent biological replicates. (A–D) Significance of differences between antibody-based therapeutic alone and antibody-based therapeutic plus tofacitinib treatments was calculated by unpaired 2-tailed t test. *P < 0.05; **P < 0.01. To determine if tofacitinib-mediated enhancement of delivery was applicable to other antibody-based therapeutics, tumor uptake of a fluorescently labeled full-length antibody was investigated. TNBCs often overexpress the EGFR and up to 90% of PDACs show EGFR overexpression. Therefore, mice with KLM-1 (Figure 2C) or MDA-MB-468 (Figure 2D) xenografts were injected with a fluorescently labeled anti-EGFR monoclonal antibody following tofacitinib or vehicle pretreatment. With vehicle-treated mice, a median of 13% of KLM-1 tumor cells, identified with an antibody against the human transferrin receptor (TfnR), were associated with a positive anti-EGFR–BV421 signal. However, with tofacitinib pretreatment, 20% of tumor cells showed positive signal (P = 0.004) (Supplemental Figure 3B). Similarly, MDA-MB-468 tumors showed an increase in antibody uptake when treated with both anti-EGFR and tofacitinib compared with tumors treated with anti-EGFR plus vehicle (9.3% to 4.0%; P = 0.04). Like the uptake of immunotoxins, tumors from mice receiving tofacitinib treatment showed a 1.5- to 2-fold increase in the number of cells exhibiting EGFR antibody binding over tumors treated with vehicle, supporting the hypothesis that improved tumor access for antibody-based therapeutics may be a broadly applicable property of tofacitinib treatment. Additionally, we chose to investigate whether tofacitinib could similarly enhance ADC delivery through evaluation of tofacitinib’s effects on the antitumor activity of anetumab ravtansine, a mesothelin-targeted ADC with reported antitumor activity in both preclinical and clinical studies against mesothelin-positive malignancies (44). The combination of tofacitinib and anetumab ravtansine in tissue culture showed no enhancement of KLM-1 killing (Figure 3A). However, in vivo, combinations with tofacitinib enhanced the antitumor effects of anetumab ravtansine, with tumor growth significantly delayed compared with anetumab ravtansine alone (P = 0.002) (Figure 3B). Treatment with anetumab ravtansine alone resulted in modest inhibition of tumor growth and a 35% increase in survival over untreated tumors (P = 0.04). As before, tofacitinib exhibited no significant antitumor activity alone. While the amount of antibody required for an observable effect was high (10 mg/kg), this dosage is comparable to published reports. Additionally, independent experiments with lower anetumab ravtansine dosing showed similar trends of enhancement, albeit with lower-magnitude effects (Supplemental Figure 4, A–D), confirming a tofacitinib-mediated enhanced delivery of this therapeutic. Thus, pretreatments of tumor-bearing mice with tofacitinib had the general effect of enhancing antibody-mediated antitumor efficacy, suggesting the existence of treatment barriers that limit the effectiveness of antibody-based therapeutics. Figure 3 Tofacitinib treatment enhances the antitumor effects of an ADC in vivo. (A) KLM-1 cells were incubated with the indicated concentration of ADC (anetumab ravtansine) and tofacitinib for 72 hours. Cell viability was measured and normalized to non–ADC-treated controls. n = 2 independent replicates performed in triplicate. (B) Mice bearing KLM-1 xenografts were treated with vehicle, tofacitinib alone, anetumab ravtansine (10 mg/kg) alone, or a combination of both (n = 5–6 mice per treatment group). Mice received 3 cycles of treatment with a week between cycles (arrows). Tumor volumes were measured and significance was calculated by unpaired 2-tailed t test between each treatment group at experimental endpoints. For KLM-1 tumors, a preliminary experiment with only 1 cycle of treatment at a lower ADC dose showed enhancement of ADC activity with tofacitinib compared with ADC alone, but of a lower magnitude (Supplemental Figure 4). (C) Kaplan-Meyer plot displaying time to death of each treatment group for mice implanted with KLM-1 tumors. Log-rank test was performed to calculate significance between anetumab ravtansine alone and anetumab ravtansine plus tofacitinib treatments. *P < 0.05; **P < 0.01; ***P < 0.001. Tofacitinib-mediated downregulation of inflammatory cytokine and arginase expression. Tofacitinib did not enhance immunotoxin activity in tissue culture, but enhanced therapeutic uptake in vivo, suggesting action at the level of the tumor itself. To determine if tofacitinib inhibition of JAK/STAT signaling could alter the TME transcriptome, RNA was isolated from KLM-1 tumors that had received no treatment, vehicle alone, or tofacitinib. Transcript profiles for murine immune-related genes were then evaluated by Nanostring technology. Transcript counts similar between vehicle and untreated tumors but altered in tofacitinib-treated tumors were identified by agglomerative clustering analysis (Supplemental Figure 5). Differential expression of this subset of genes was then determined for vehicle- and tofacitinib-treated tumors, and genes with at least 2-fold downregulation identified (Table 1). Tofacitinib-downregulated transcripts could be broadly grouped into 3 functional categories: neutrophil/granulocyte chemokines, monocyte chemokines, and B cell activation proteins. The transcripts showing the greatest reductions in tofacitinib-treated tumors were neutrophil chemokines Cxcl2 (–7.46-fold) and Cxcl3 (–6.29-fold), with monocyte chemokines (Ccl4, –3.62-fold; Ccl1, –3.35-fold, Ccl3, –2.86-fold) and B cell activation pathway components (Pax5, –3.78-fold; Il13, –2.48-fold) also reduced (Table 1). Transcripts for arginase-1 (–2.37-fold) and cyclooxygenase 2 (COX-2) (–1.58-fold), which are often highly expressed in MDSCs and suppress antitumor immune activity, were also downregulated. Fewer genes were upregulated in tofacitinib-treated KLM-1 tumors compared with vehicle/untreated tumors (Table 2). The most differentially expressed gene was that encoding interferon-α (+3.85-fold), an antiviral cytokine that stimulates natural killer (NK) cell cytotoxic activity upon STAT1 inhibition (50). However, genes upregulated by tofacitinib treatment did not obviously cluster into functional groupings and were not studied further. Table 1 Mouse genes showing greater than 2-fold downregulation in mRNA transcript levels after tofacitinib treatment (T) compared with vehicle-treated (V) mice Table 2 Mouse genes showing greater than 2-fold upregulation in mRNA transcript levels after tofacitinib treatment (T) compared with vehicle-treated (V) mice Nanostring analysis revealed that tofacitinib treatment showed the largest effect on transcription of neutrophil and monocyte chemoattractants. To determine if transcript changes correlated with protein levels, CXCL2 and CCL4 serum levels were analyzed by ELISA. Furthermore, circulating IL-6 and CCL2 levels were analyzed because these cytokines are routinely lowered in RA patients treated with tofacitinib (42, 51). Non–tumor-bearing mice displayed low levels of circulating CXCL2 (15.4 pg/ml) and essentially undetectable levels of circulating CCL4 (2.3 pg/ml). Tumor-bearing mice showed upregulation of both chemokines (CXCL2, 61.8 pg/ml; CCL4, 38.4 pg/ml), indicating that the tumor had stimulated production (Figure 4A). Treatment with tofacitinib significantly reduced serum levels of both chemokines, decreasing CXCL2 to 25.1 pg/ml, only a 1.6-fold increase over naive mice (P < 0.001). Circulating CCL4 was similarly reduced, from 38.4 pg/ml in vehicle-treated tumors to 8.4 pg/ml in tofacitinib-treated mice (P = 0.02). Naive mice showed slightly higher levels of CCL2 compared with the other chemokines (30.6 pg/ml); however, in mice bearing KLM-1 tumors, circulating CCL2 doubled to 61 pg/ml. Treatment with tofacitinib reduced this expression to 38.9 pg/ml (P = 0.004). Mice bearing KLM-1 tumors showed a minimal increase in IL-6 over naive mice (33.6 to 38.1 pg/ml), which was abrogated in tofacitinib-treated mice (32.7 pg/ml) (Figure 4B). However, mice with MDA-MB-468 tumors showed a tripling of circulating IL-6 compared with naive mice (33.6 to 87.1 pg/ml), consistent with the role that IL-6 plays in TNBC growth and metastasis. Treatment with tofacitinib resulted in a significant decrease in IL-6, to levels comparable to naive mice (32.8 pg/ml) (P = 0.01). These data confirmed that tofacitinib reduced key cytokines associated with inflammatory cell recruitment and tumorigenesis. Figure 4 Tofacitinib treatment reduces the levels of circulating proinflammatory chemokines and tumor-associated arginase. (A) ELISA quantifying murine CCL4, CXCL2, and CCL2 in serum from mice with KLM-1 xenografts treated with vehicle or tofacitinib. Also shown are chemokine levels for non–tumor-bearing mice (naive). Results are from at least 3 independent animals for each treatment condition. (B) ELISA quantifying murine IL-6 in serum from mice with KLM-1 (left) or MDA-MB-468 (right) xenografts treated with vehicle or tofacitinib. Also shown are IL-6 levels from non–tumor-bearing mice. Results are from at least 2 independent animals for each treatment with assays repeated in duplicate. (C) Arginase activity was measured in tofacitinib- or vehicle-treated KLM-1 (left) or MDA-MB-468 (right) tumors by incubating supernatants from tumors with L-arginine. Enzymatic activity was determined through colorimetric measurement of urea generated by arginase hydrolysis of L-arginine. One AU of arginase activity was defined as the amount of enzymatic activity required for production of 1 μg urea in 1 hour. *P < 0.05; **P < 0.01; ***P < 0.001 by 2-tailed t test comparing vehicle- and tofacitinib-treated animals (A and B) or tumors (C). The presence of TANs, MDSCs, and TAMs is linked frequently to a tumor-permissive environment through release of proinflammatory cytokines and production of immune-suppressing molecules like arginase-1 or COX-2 (52–54). As arginase expression is regulated by STAT signaling (55) and showed more than 2-fold downregulation of the transcript level upon tofacitinib treatment (Table 1), changes in arginase activity levels within tumors were also examined. The enzymatic activity of arginase-1 in tumor supernatants was measured through the conversion of L-arginine to urea (56). Tofacitinib treatment of KLM-1 xenografts showed an approximately 20% reduction in tumor-associated arginase activity compared with vehicle-treated tumors (P = 0.02) (Figure 4C). Treatment of MDA-MB-468 xenografts with tofacitinib showed a similar trend, significantly reducing tumor-associated arginase enzymatic activity to 43% of that of vehicle-treated tumors (P = 0.03) (Figure 4C). Taken together, these data suggested that tofacitinib alters the TME by reducing the recruitment and/or activity of certain immune populations. Tofacitinib reduces recruitment of TANs, TAMs, and myeloid cells. To determine whether tofacitinib treatment altered the composition of the tumor, mice were treated with vehicle or tofacitinib (Figure 5A). Given the depletion of myeloid cell and neutrophil chemotactic proteins, antibodies reactive for markers of myeloid cells (α-CD11b or α-GR-1), neutrophils (α-myeloperoxidase [α-MPO] or α-LY6G), or murine macrophages (α-F4/80) were utilized to determine if these inflammatory cell populations within the tumor were altered by tofacitinib treatment. Images were captured for each treatment group and scored for the presence and quantity of each cell marker from at least 20 random fields for each treatment. In KLM-1 tumors, TAMs were obviously depleted in tofacitinib-treated animals compared with vehicle-treated tumors (Figure 5A). Vehicle-treated tumors showed a median of 42 F4/80+ cells per field (range, 7–140), which was significantly reduced in tofacitinib-treated animals to 18 cells/field (range, 6–88) (P < 0.01). Tumor-associated CD11b+ myeloid cells also decreased in response to tofacitinib treatment (Figure 5B). Vehicle-treated tumors showed CD11b+ cells scattered throughout the tumor stroma, while these cells were barely detectable in tofacitinib-treated tumors. Vehicle-treated tumors scored a median of 20 CD11b+ cells per field (range, 3–45), while treatment with tofacitinib significantly reduced this number to 1 cell/field (range, 0–8) (P < 0.001). Similarly, vehicle-treated tumors showed MPO+ cells (arrowheads) within the stroma, but also among the tumor cells (Figure 5C). The population of MPO+ cells was also altered significantly in tofacitinib-treated tumors, as MPO+ cells decreased by approximately 80% compared with vehicle, from a median of 20 MPO+ cells/field of view (range, 2–88) to 3 MPO+ cells per field (range, 0–47) (P < 0.001) (Figure 5C). Finally, to confirm that TANs were depleted by tofacitinib, a second granulocyte marker, LY6G, was examined (Figure 5D). Vehicle-treated tumors showed a median of 17 LY6G+ cells/field of view (range, 0–104), again in both the tumor stroma and among the malignant cells. Tofacitinib significantly reduced LY6G+ cells to a median of merely 1/field (range, 0–41). Figure 5 Treatment with tofacitinib reduced the number of inflammatory cells within the tumor microenvironment. (A) Representative images of F4/80+ macrophage populations in KLM-1 xenografts after treatment with vehicle or tofacitinib. Original magnification, ×320. F4/80+ macrophages were counted from at least 20 randomly imaged fields, with counts from each field shown with the median for each treatment. (B) Representative images of CD11b+ myeloid populations in KLM-1 xenografts treated with vehicle or tofacitinib. Cells staining for CD11b were counted from at least 20 randomly imaged fields, with counts from each field shown with the median for each treatment. (C) Representative images of cell populations stained for myeloperoxidase (MPO) in KLM-1 xenografts after treatment with vehicle or tofacitinib. MPO+ cells (arrowheads) were counted from at least 20 randomly selected fields, with counts from each field shown with the median for each treatment. (D) Representative images of LY6G+ granulocyte populations in vehicle- or tofacitinib-treated KLM-1 xenografts. LY6G+ cells were counted from at least 20 randomly selected fields, with counts from each field shown with the median for each treatment. **P < 0.01; ***P < 0.001 by unpaired 2-tailed t test (A–D). Similar trends were noted for IHC staining of inflammatory cell populations in MDA-MB-468 tumors (Supplemental Figure 6A), although there were fewer inflammatory cells overall. Tofacitinib treatment modestly, but significantly, reduced median CD11b+ cells compared with vehicle from 9 (range, 0–60) to 4 (range, 0–28) (Supplemental Figure 6B, P = 0.009). As the CD11b+ staining in MDA-MB-468 tumors was less well defined than in KLM-1 tumors, we also identified myeloid cells using the GR1-LY6G marker. Tofacitinib similarly resulted in a moderate reduction in tumor-associated GR1-LY6G+ cells from a median of 16 cells/field (range, 5–46) to 12 (range, 0–32) (P = 0.04) (Supplemental Figure 6C). Finally, tofacitinib depleted MPO+ cells in MDA-MB-468 tumors (Supplemental Figure 6D). Vehicle-treated tumors showed a median of 18 MPO+ cells/field (range, 0–41), while tofacitinib-treated tumors only had 8 MPO+ cells/field (range, 0–46) (P = 0.04). In the above experiments, tumors were harvested 3 days after receiving the first injection of tofacitinib and then scored for the presence of short-lived inflammatory cells: neutrophils (typical t1/2 of less than a day), monocytes (t1/2 of 1–2 days), and TAMs, which were substantially lowered after 3 days treatment with other macrophage-depleting agents. Our data confirm that short-duration treatments can result in tofacitinib-mediated depletion of chemokine levels and reduced recruitment of inflammatory cells into the TME. Tofacitinib treatment reduces tumor-associated inflammatory cells in a syngeneic breast cancer model. Xenograft tumor models allow therapeutic compounds to be evaluated against human cancers in a living animal. However, the absence of effector T cells does not permit an adaptive immune response. To investigate tofacitinib activity in an animal with an intact immune system, a syngeneic tumor model of breast cancer was evaluated. 4T-1 mammary carcinoma cells were implanted in the mammary pad of female BALB/c mice and treated with tofacitinib or vehicle, and IHC was performed to assess the effect of tofacitinib on cell recruitment into the TME (Supplemental Figure 7, A–D). Similar to results in the xenograft models, tumor-associated inflammatory cells were reduced following tofacitinib treatment, with significant decreases noted in the number of CD11b+ cells (P = 0.04), MPO+ cells (P < 0.001), LY6G+ granulocytes (P < 0.001), and F4/80+ macrophages (P = 0.002). However, as seen in both xenograft tumor models, no significant differences in tumor growth rate were recorded with tofacitinib treatments, suggesting that antitumor cell functions that otherwise might be keeping the tumor in check were not significantly compromised under these short-duration treatment regimens. (Supplemental Figure 7E). Arginase enzymatic activity within the syngeneic 4T-1 breast cancer tumors was also measured as before, with tofacitinib treatment reducing tumor-associated arginase activity to 68% of that of vehicle-treated tumors (Supplemental Figure 7F, P < 0.001). Non-target uptake of immunotoxins by tumor-associated inflammatory cells. The reduction in tumor-associated inflammatory cells following tofacitinib treatments coupled with enhanced uptake of antibodies into malignant cells suggested that inflammatory cells could be altering therapeutic protein availability. To address this, we first investigated whether tofacitinib altered the pharmacokinetics of immunotoxin metabolism. Sera from mice bearing KLM-1 tumors pretreated with tofacitinib or vehicle were harvested at 0, 15, 30, 45, and 60 minutes after injection and assayed for LMB-100 immunotoxin levels by ELISA (Figure 6A). Mice treated with vehicle had an immunotoxin half-life of 25 minutes, similar to previous reports (57), while mice treated with tofacitinib had a half-life of 46 minutes (P = 0.02). Tofacitinib additionally increased the immunotoxin exposure (area under the curve from t = 0 to t = 60) by 35%, suggesting that malignant cells had a greater exposure to immunotoxin molecules. Likewise, drug levels for the immunotoxin HB21-PE40 were analyzed kinetically in mice bearing MDA-MB-468 tumors, with vehicle-treated tumors showing an immunotoxin half-life of 15 minutes, while mice treated with tofacitinib had a half-life of 30 minutes (Supplemental Figure 8A, P = 0.04). These data confirmed that tofacitinib increased the amount of immunotoxin available to interact with malignant cells, potentially by reducing inflammatory cell clearance of immunotoxin, minimally at the tumor itself, but possibly at other locations. To determine if tofacitinib acted mainly on inflammatory cell populations stimulated by the presence of a malignancy, or on inflammatory cells in general, a similar experiment was performed in naive mice. As before, sera from mice pretreated with tofacitinib or vehicle and injected with LMB-100 were harvested at 0, 15, 30, 45, and 60 minutes after injection and assayed for LMB-100 immunotoxin levels by ELISA (Figure 6B). Naive mice showed an LMB-100 half-life of 23 minutes, while tofacitinib-treated mice had an increased half-life of 32 minutes (P = 0.66). Treatment with tofacitinib increased immunotoxin exposure by almost one-third compared with vehicle-treated mice, suggesting that tofacitinib may be acting more generally and not just on tumor-associated inflammatory cells. Figure 6 Immunotoxin metabolism with and without tofacitinib treatments. (A) Mice bearing KLM-1 tumors were treated with vehicle or tofacitinib at t = –48 and –24 hours. At t = 0, mice were injected with LMB-100 and blood was harvested at t = 0, 15, 30, 45, and 60 minutes. Circulating LMB-100 was detected by ELISA and normalized to a standard curve for purified LMB-100. The half-life and area under the curve were calculated from best-fit regression analysis. Results are averaged from 2 biological replicates. (B) Naive mice were treated with vehicle or tofacitinib at t = –48 and –24 hours. At t = 0, mice were injected with LMB-100 and blood was harvested at t = 0, 15, 30, 45, and 60 minutes. Circulating LMB-100 was detected by ELISA and normalized to a standard curve for purified LMB-100. The half-life and area under the curve were calculated from best-fit regression analysis. Results are averaged from 4 biological replicates. (C) Mice bearing KLM-1 xenografts were pretreated with either vehicle or tofacitinib, then administered 50 μg LMB-100–A647 for 3 hours. Dissociated tumors were scored for the presence of LMB-100–A647 in malignant cells and inflammatory populations. Tumor cells were identified by staining with anti-huEGFR, and tumor-associated inflammatory cell populations were identified by staining with anti-mCD11b. The percentage of huEGFR or mCD11b cells containing immunotoxin in vehicle or tofacitinib-treated mice is shown. n = 4 biological replicates. (D) CD11b+ cells containing LMB-100–A647 were further discriminated into CD68+ (macrophage) or LY6G+ (neutrophil) populations. The percentage of tumor-associated LMB-100–A647+ cells displaying each cell marker is shown. n = 4 biological replicates. *P < 0.05; **P < 0.01; ***P < 0.001 by unpaired 2-tailed t test (A–D). ns, not significant. If antibodies targeted to malignant cells are ingested or otherwise depleted by inflammatory cells, then peritumoral scavenging may result in less effective therapies. To address this specifically, the cellular fate of fluorescently labeled immunotoxin in tumor-bearing mice was further investigated; immunotoxins rather than full-length antibodies were chosen to both avoid potentially confounding results with Fc receptor–mediated uptake, as well as to stay consistent with our previous antitumor studies. KLM-1 tumors from mice pretreated with tofacitinib or vehicle and injected with LMB-100-A647 were dissociated and stained with anti–human EGFR (anti-huEGFR) to identify malignant cells and anti-mCD11b to identify murine (m) myeloid populations. mCD11b+ cells were counterstained with antibodies against mCD68 or mLY6G to distinguish macrophage and granulocyte populations, respectively. Cells containing fluorescent immunotoxin were identified by flow cytometry and separated into huEGFR+ or mCD11b+ populations, with mCD11b+ cells further divided into mCD11b+mCD68+ (macrophage) or mCD11b+mLY6G+ (granulocyte) populations (Figure 6C). Almost two-thirds of immunotoxin-positive cells in vehicle-treated tumors were mCD11b+, suggesting that the majority of immunotoxin uptake was associated with off-target inflammatory cell populations (Figure 6C). Tofacitinib-treated tumors still showed a majority of immunotoxin within mCD11b+ cells; however, this population was significantly reduced compared with vehicle-treated tumors (55%, P = 0.007). When this population was further divided into mCD11b+mCD68+ macrophage and mCD11b+mLY6G+ granulocyte populations, tofacitinib-treated tumors showed a one-third reduction in macrophage-associated LMB-100–A647 (P = 0.05), but only a minimal reduction in granulocyte-associated immunotoxin (Figure 6D). Concurrent with the reduction in mCD11b+LMB100–A647+ cells in tofacitinib-treated tumors was an increase in huEGFR+LMB100–A647+ cells compared with vehicle-treated tumors (tofacitinib, 34% vs. 22% vehicle; P < 0.001). In tumor-bearing animals, inflammation is not confined to the TME but also noted in the spleen, a significant source of precursor inflammatory cells which transit to the tumor, differentiate into TAMs/TANs, and accumulate within the tumor (58, 59). Therefore, we examined spleens from tumor-bearing mice with and without tofacitinib treatment. In mice bearing KLM-1 tumors, we noted a 25% increase in spleen weight (0.22 ± 0.02 g, n = 4) compared with naive mice (0.16 ± 0.01 g, n = 2). In contrast, spleens from tumor-bearing mice receiving tofacitinib showed only 10% increase in weight (0.18 ± 0.01 g, n = 4) over naive mice, suggesting a drug-mediated reduction in splenic inflammation, consistent with published reports (60). Further, when splenocytes from naive and tumor-bearing mice were analyzed by flow cytometry, tofacitinib treatment resulted in a 10% reduction in overall CD11b+ populations and a 20% reduction in CD68+ populations (Supplemental Figure 8, B and C). Thus, it is likely that short-duration tofacitinib treatments reduce potential non-target sinks for antibody-based agents not only at the tumor site, but at other locations where inflammatory cells are prone to accumulate. Together, these data suggest that tumors with high levels of phagocytic inflammatory cells may divert a significant proportion of anti-cancer antibodies and that reducing their presence in and around a tumor could significantly improve the amount of therapeutic antibody available for interacting with target malignant cells. Discussion Previously, we reported that the generation of anti-immunotoxin antibodies in immunocompetent mice was inhibited by tofacitinib, reducing titers by more than 200-fold (41). Here, we extend those findings and report on a second substantial benefit of using tofacitinib, namely the increased delivery of antibody therapeutics to malignant cells, achieved by reducing cytokine signaling and the recruitment of tumor-associated inflammatory cells. Consistent with enhanced delivery, we noted increased immunotoxin efficacy in TNBC and PDAC xenograft tumor models. Expanding on these initial experiments, we demonstrated that tofacitinib also enhanced the activity of an ADC targeting mesothelin in a pancreatic tumor xenograft. In experiments designed to quantify delivery to malignant cells, tofacitinib pretreatment resulted in greater uptake of antibody-based agents into malignant cells and an increased immunotoxin half-life in tofacitinib-treated mice. While tofacitinib-mediated reductions of inflammatory cells in general was not a surprise considering its reported antiinflammatory activity in the joints of rodent models (43) and human RA patients (45), decreased inflammation within tumors and decreased non-target uptake of immunotoxin by inflammatory cells was not anticipated. Our data suggest that depletion of these cell types may explain the in vivo enhancement of antibody-based therapeutics in several xenograft models. The chronic inflammatory state associated with many cancers often results in the accumulation of innate immune cells associated with wound healing and repair, which respond to cytokines released by the tumor and in turn, release growth factors, angiogenic factors, and cytokines that promote tumor growth, metastases, and immune suppression (61). This polarization of the TME towards an alternative state is predicated on cytokine crosstalk between tumor cells, stroma, and the host immune system, suggesting that targeting common signaling pathways could disrupt a protumorigenic environment (24, 62, 63). Intriguingly, a recent article reported on systemic inflammation with monocyte and neutrophil mobilization that could be abolished by a few treatments with antiinflammatory agents (64). In our study, histological examination of tumor inflammatory cell populations confirmed that TAM and TAN populations were depleted in tofacitinib-treated tumors. Published reports indicate that the proliferation of TAMs within the TME is diminished compared with counterparts in normal tissue; similarly, neutrophils are short-lived cells and rapidly undergo apoptotic cell death, so both inflammatory populations must be continuously recruited to tumors via chemotactic pathways (2). Accumulation of TAMs is an independent predictor of poor patient outcomes in many cancer types, including breast and pancreatic cancers, and are associated with increased resistance to therapeutics (65, 66). Additional depletion of neutrophils in a cancer setting may play a beneficial role, as TANs are associated with disease progression and poor outcomes in many cancers (67–69). Similarly, the modulation of tumor-supporting immune cell populations has been recently identified as a significant component in the effectiveness of cancer treatments with antibodies against immune checkpoint proteins PD-1/PD-L1 (70), and CTLA-4 (71). Reductions in inflammatory cells after tofacitinib treatment in the 4T-1 syngeneic breast cancer model confirmed that our findings were not peculiar to human xenografts or athymic nude mice. In fact, short-term treatments with tofacitinib achieved qualitative effects similar to other agents designed to inhibit macrophage or monocyte recruitment to tumors, such as novelizumab, carlumab, or RG7155 (2). In PDAC and other tumor models, various anti-macrophage treatments have increased the susceptibility of malignant cells to either chemotherapy or checkpoint inhibitors (72–74), and the use of antiinflammatory drugs in combination with the standard of care in breast (75) and pancreatic (76) cancers has shown some promise. In addition to these combinations, we propose a novel use for agents that inhibit the recruitment of inflammatory cells, namely the increased bioavailability and enhanced uptake of antibodies into malignant cells. In support of this, we report substantial non-target uptake of immunotoxins by inflammatory cells in tumor-bearing mice. Because of the short half-lives of recombinant immunotoxins, uptake was measured 3 hours after injection. Results indicated that at this time point, a majority of tumor-associated immunotoxin was recovered within inflammatory cells in vehicle-treated mice. In tofacitinib-treated mice, there were fewer inflammatory cells containing immunotoxin, and thus we speculate that decreased non-target uptake leads to increased on-target delivery to malignant cells. While these experiments provided a quantitative insight into the short-term fate of immunotoxins, long-term effects were also evident, seen in the enhanced antitumor activity observed with 3 unique antibody-based therapeutic agents. Clinical evidence suggests that depletion of tumor-associated suppressive cell populations may play a role in enhancing multiple classes of anticancer therapies (77). Gemcitabine specifically induces apoptosis in MDSC populations and depletes neutrophils in human patients (78, 79), while having no effect on T cells, B cells, NK cells, or dendritic cells. Combination of gemcitabine with immunotherapies has proven significantly more effective than immunotherapy or gemcitabine alone, suggesting that depletion of MDSC or neutrophil populations may be a general mechanism to improve patient outcomes (80). Relevant to our study, treatment with gemcitabine in conjunction with immunotoxin resulted in better tumor clearance in vivo than either agent alone, while having no enhancement effect in vitro (81). These studies suggest that compounds that inhibit the expansion or recruitment of immunosuppressive and tumor-promoting immune populations may prove effective at enhancing antibody-based therapies. However, it remains difficult to identify these agents using in vitro screening because contributions of the TME are difficult to duplicate outside the intact animal. Previous efforts for improving drug delivery were primarily predicated upon 2 different approaches: normalization of the vasculature through antiangiogenesis agents, or depletion of ECM and tumor stromal cells (82). However, recent focus has shifted to reducing the inflammatory state of the tumor itself to stimulate vascular normalization, reduce edema, and deplete protumorigenic inflammatory cells (7). While our study provides evidence that reducing tumor inflammation may enhance the efficacy of tumor-directed antibodies, it has not revealed whether increased antibody delivery reflects only relief from nonspecific uptake by inflammatory cells or if other mechanisms are operative. We have yet to examine whether tofacitinib affects normalization of endothelial vessel organization (83), decreases tumor interstitial pressure due to reduction in inflammation, or reduces matrix proteins that can inhibit therapeutic antibody penetration (84). However, as tofacitinib is approved for treatment of fibrosis and significantly decreases deposition of ECM proteins, especially collagens (85), this may be an additional benefit of using this specific compound. By microscopy, some tofacitinib-mediated reduction in fibrosis within our tumor models was noted, but this was not quantified. Further, when monitoring antitumor effects, possible contributions from reduced levels of cytokines promoting tumor survival or proliferation cannot be excluded. Future studies will focus on these important issues. Of additional interest, CD11b+ populations are inversely correlated with the effectiveness of checkpoint inhibitor treatments (86, 87). A potential role for tofacitinib in this setting is especially intriguing, as not only does tofacitinib deplete tumor-associated CD11b+ populations, it also suppresses transcription of PD-L1 through a STAT-dependent mechanism (88). As shown in Table 1, we observed a decrease in transcript levels of PD-L1 in the tumors of tofacitinib-treated mice. In conjunction with the observed improvement in tumor access for antibody-based therapeutics, we speculate that these effects could result in tofacitinib enhancement of checkpoint-based immunotherapy agents in poorly responsive cancers, including pancreatic cancers. However, it is unclear how tofacitinib might affect T cell populations required for effective tumor clearance upon checkpoint blockade. In summary, our data show that tofacitinib, an FDA-approved small-molecule JAK/STAT inhibitor with an acceptable safety profile in humans, alters the TME through suppression of the chemokines that attract inflammatory cells. Broadly, our data suggest that depletion of tumor-associated inflammatory cells through inhibition of chemokine signaling may improve the treatment efficacy of antibody-based anticancer therapeutics and suggest that tofacitinib or similar antiinflammatory drugs could be used in combination with antibody-based therapeutics. Methods Cell culture. The KLM-1 (PDAC) cell line was derived by Kimura et al. from a liver metastatic lesion (89). The MDA-MB-468 (TNBC) cell line was obtained from the American Type Culture Collection (HTB-132). MDA-MB-468 cells were cultured in DMEM supplemented with 10% heat-inactivated FBS, Glutamax, nonessential amino acids (NEAAs), and sodium pyruvate. KLM-1 cells were cultured in ATCC-formulated RPMI 1640 medium (Thermo Fischer Scientific) with Glutamax, NEAAs, and 10% heat-inactivated FBS. Cellular viability. Cells were plated in 96-well format at 10,000 cells/well. After 24 hours, recombinant immunotoxin or ADC was added at the concentration indicated with 0.1–16 μM tofacitinib or DMSO control as indicated. After 72 hours, viability was determined with the CellTiter-Glo Viability Assay Kit (Promega). Data are presented as a percentage of control cells. Data are from at least 2 independent replicate experiments in triplicate. Cancer xenograft models. MDA-MB-468 or KLM-1 tumors were established in 6- to 9-week-old female nude athymic mice (Charles River Laboratories). After tumor volume had reached approximately 100 mm3, mice were randomized into treatment groups (n = 6–8 mice per group) and treated with vehicle alone, tofacitinib (5 mg/kg, twice), HB21-PE40 (0.1 mg/kg), or LMB-100 (2.5 mg/kg), or the combination of tofacitinib and immunotoxin. Mice were treated every other day for 1 (MDA-MB-468) or 2 (KLM-1) treatment cycles with 4 injections per cycle. Tofacitinib was dissolved in 30% PEG300 and 5% Tween 80 in PBS and injected i.p. once in the morning with the second injection 8 hours later to mimic the twice-daily human dosing schedules. Immunotoxin injections were performed every other day via tail vein for each mouse in conjunction with the second daily tofacitinib dosing. Anetumab ravtansine (provided by Bayer AG) was diluted in the supplied vehicle (10 mM histidine, 130 mM glycine, 5% sucrose in water). Each treatment cycle consisted of 10 mg/kg tofacitinib (3 times) and 10 mg/kg anetumab ravtansine (once), with mice treated once per day with tofacitinib and anetumab ravtansine delivered via tail vein concurrent with the third dose. Tumor volume and animal weight were measured at least 3 times weekly. Tumor volume was calculated as 0.5 × (l × w2). Animals were euthanized by CO2 inhalation once tumors reached 1,200 mm3, became necrotic, or animals became moribund. Time to endpoint for each mouse was displayed on a Kaplan-Meyer plot and statistical significance calculated by log-rank test. Syngeneic tumor model. 4T-1 tumors were established in 6- to 9-week-old female BALB/c mice (Charles River Laboratories). After tumor volume had reached approximately 200 mm3, mice were randomized into treatment groups (n = 6–8 mice per group) and were left untreated, treated with vehicle alone, or tofacitinib (5 mg/kg, twice). Mice received 4 treatments every other day and were euthanized once tumors reached 1,200 mm3. Nanostring analysis. Total RNA was purified from homogenized KLM-1 tumors 24 hours after treatment with 2 cycles of vehicle or tofacitinib (1 cycle: 5 mg/kg tofacitinib twice at 8-hour intervals, then a 24-hour rest period) using the QIAGEN RNeasy RNA Purification kit. RNA from untreated, vehicle-treated, or tofacitinib-treated tumors (n = 3 tumors per treatment group) was loaded onto the Nanostring nCounter PanCancer Immune Profiling Panel (Nanostring Technologies Inc) according to manufacturer’s specifications. mRNA transcript counts were analyzed using NSolver software (Nanostring Technologies), with differential expression compared by pooled cluster analysis. Transcript differences between vehicle and tofacitinib-treated RNA pools were ordered according to the magnitude of fold change and transcripts with greater than 2-fold changes are displayed in Tables 1 and 2. ELISA. Cytokine levels were determined by testing serum from tofacitinib-treated, vehicle-treated, or untreated tumor-bearing mice using the Mouse IL-6 Quantikine ELISA (M6000B), Mouse CCL2/JE/MCP-1 Quantikine ELISA (MJE00), Mouse CXCL2/MIP-2 Quantikine ELISA Kit (MM200), Mouse CCL4/MIP-1 beta Quantikine ELISA Kit (MMB00), or Mouse CCL3/MIP-1 alpha Quantikine ELISA (MMA00) (all R&D Systems). Results were compared to cytokine levels detectable in serum from naive mice and significance was assessed using unpaired 2-tailed t tests. Tumor uptake and flow cytometry. Mice were pretreated with 2 cycles of vehicle or tofacitinib (1 cycle: 5 mg/kg twice at 8-hour intervals, then a 24-hour rest period) and then injected via tail vein with 50 μg of Alexa Fluor–labeled immunotoxin or 10 μg fluorescently labeled antibody. After 3 hours, tumors were harvested, minced, and resuspended in HBSS supplemented with Liberase TL (0.2 U/ml) (Roche Life Sciences) and DNAse (0.1 mg/ml) (Sigma-Aldrich) at 3°C for 45 minutes. The dissociated tumor mix was passed through a 70-μM cell strainer, single cells were pelleted in HBSS plus 10% FBS, and immediately used for downstream applications or stored in tissue culture media at –80°C for future analysis. For cytometric analyses, single-cell suspensions were enumerated and 1 × 106 cells for each sample were blocked in 4% serum, 2% BSA, and 1% mouse FcBlock (BD Biosciences) for 30 minutes. Anti–mouse CD11b-FITC (catalog 557396) or CD11b-BV421 (catalog 562605), CD68-PE (catalog 566386) or CD68-BV421 (catalog 566388), and LY6G-PE (catalog 551461) (BD Biosciences) were used to identify murine hematopoietic populations. Anti-huEGFR (BD Biosciences) was used to identify malignant cells. EGFR+ human cells were screened for the presence of immunotoxin, and the percentage of human cells with immunotoxin was compared for mice treated with vehicle plus immunotoxin or tofacitinib plus immunotoxin. Alternatively, the percentage of dissociated tumor cells staining EGFR+ and LMB-100–A647+ was compared to the percentage of CD11b+immunotoxin+ or CD11b+CD68+immunotoxin+ or CD11b+LY6G+immunotoxin+ cells for each mouse. Compensation controls were performed for each fluorophore pairing. Isotype antibodies for each fluorophore were used to set background staining levels. Cells were assayed on a FACSCanto II flow cytometer (BD Biosciences). Data were analyzed in FlowJo v10 software (FlowJo, LLC). Alexa Fluor labeling of proteins. Immunotoxins or antibodies were labeled with the Alexa Fluor-647 Protein Labeling Kit (Thermo Fisher Scientific). Labeled proteins were purified on PD-10 desalting columns (GE Life Sciences) and concentrations determined by spectrophotometry. The dye/protein ratio was measured according to the manufacturer’s instructions and was within the manufacturer-specified optimal range. To ensure that the fluorescence-labeling process did not interfere with immunotoxin activity, in vitro cell killing assays were performed comparing labeled and unlabeled immunotoxin. Less than 1-log reduction in cell killing was noted between the 2 preparations at equivalent concentrations, indicating that the labeled immunotoxin was still capable of binding and internalization. IHC. Formalin-fixed xenografts were embe 

Looking for a manufacturing partner for your antibody drug conjugates (ADCs)? Novasep has been a leading contract service provider in the ADC arena for more than 10 years. Via Krishan Maggon
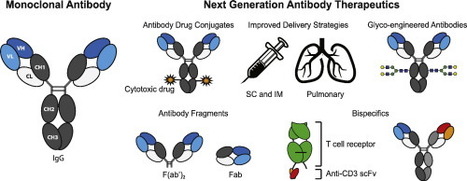
The state-of-play and future of antibody therapeutics #mAbs #ADCs #multispecifucs #biosimilars #biobetters https://t.co/tXTNIurbeE It has been over four decades since the development of monoclonal antibodies (mAbs) using a hybridoma cell line was first reported. Since then more than thirty therapeutic antibodies have been marketed, mostly as oncology, autoimmune and inflammatory therapeutics. While antibodies are very efficient, their cost-effectiveness has always been discussed owing to their high costs, accumulating to more than one billion dollars from preclinical development through to market approval. Because of this, therapeutic antibodies are inaccessible to some patients in both developed and developing countries. The growing interest in biosimilar antibodies as affordable versions of therapeutic antibodies may provide alternative treatment options as well potentially decreasing costs. As certain markets begin to capitalize on this opportunity, regulatory authorities continue to refine the requirements for demonstrating quality, efficacy and safety of biosimilar compared to originator products. In addition to biosimilars, innovations in antibody engineering are providing the opportunity to design biobetter antibodies with improved properties to maximize efficacy. Enhancing effector function, antibody drug conjugates (ADC) or targeting multiple disease pathways via multi-specific antibodies are being explored. The manufacturing process of antibodies is also moving forward with advancements relating to host cell production and purification processes. Studies into the physical and chemical degradation pathways of antibodies are contributing to the design of more stable proteins guided by computational tools. Moreover, the delivery and pharmacokinetics of antibody-based therapeutics are improving as optimized formulations are pursued through the implementation of recent innovations in the field. Via Krishan Maggon

Krishan Maggon 's curator insight,
March 6, 2017 3:11 AM
Advanced Drug Delivery Reviews Available online 2 December 2016 In Press, Corrected Proof
Starting in February 2016, ADC Review / Journal of Antibody-drug Conjugates will offer select (video) interviews with KOLs (Key Opinion Leaders) and experts ...
Antibody-drug conjugates (ADCs) represent a rapidly growing class of anti-cancer therapeutics in which a potent small molecule is linked to a monoclonal ...

Novasep, a leading player in the life sciences services industry, recently announced that it will expand its presence in the Antibody Drug Conjugate (ADC) market.
As of today, there are a total of 367 monoclonal antibodies either on the market or in development. 124 are fully human, 146 are humanized, 37 are chimeric, and 56 are murine, or mice antibodies. Of these 367, 38 are FDA approved since the first got through in 1986. 16 of those 38 have been approved in the last 5 years alone, which translates to 42%. We are gaining traction quickly, it seems.
Of the 38 approved mabs, 18 are humanized, 14 are fully human, 7 are chimeric, and only one is murine. The difference between the types is this: A chimeric mab is a cross between an animal and a human antibody engineered to react in a human setting. Humanized means that the antibody is mostly converted into a human form but originally came from an animal, usually a mouse. The term "fully human antibody" though is actually a bit misleading because in almost all cases it is not actually taken from a human. It is generally taken from a mechanism called a phage display, where human antibody genes are injected into a phage virus, which then expresses the antibodies on its coat. The best ones are then harvested from the virus. Think of it as microbiological agriculture.
Zooming back out to mabs in general, the top 10 bestselling mabs in 2014 account for $68B of the $78B total mab market last year. Number 1 is the fully human adalimumab, or Humira for rheumatoid arthritis, which looks set to eventually overtake Lipitor as the biggest blockbuster drug of all time. Humira was discovered via phage display, along with (nearly) all other fully human mabs on the market or in development. Via Krishan Maggon
Gilbert C FAURE's insight:
thks for the economical actualizedoverview
Krishan Maggon 's curator insight,
June 12, 2015 1:46 AM
Humira was the best selling human medicinal brand in 2014 but will be overtaken by Gilead Hepatitis C antiviral sofosbuvir and Harvoni.

Blood Cancer Journal is a peer-reviewed, open access online journal publishing pre-clinical and clinical work in the field of hematology with ramifications into translational biology research down to new therapies...
From
www

Safety and activity of the anti-CD79B antibody–drug conjugate polatuzumab vedotin in relapsed or refractory B-cell non-Hodgkin lymphoma and chronic lymphocytic leukaemia: a phase 1 study - The Lanc...
Fierce 15 member Mersana Therapeutics, a developer of customizable next-generation antibody-drug conjugates, announced that its lead anticancer candidate showed promise in preclinical studies. Via Krishan Maggon
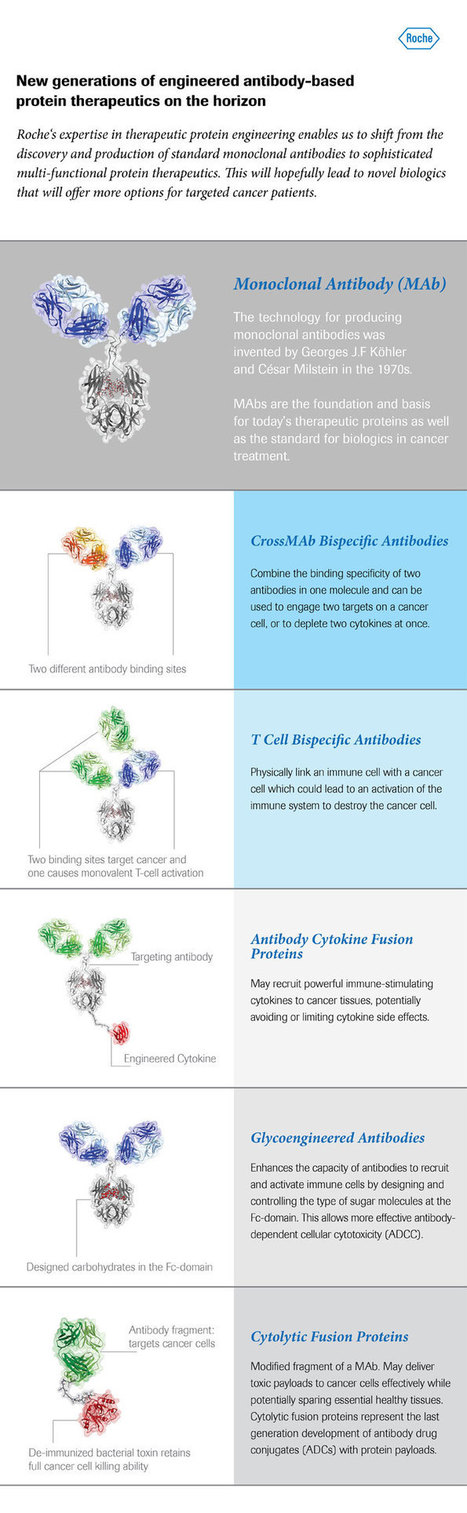
Roche currently has several different therapeutic antibody technologies in early clinical development which in the future could hopefully be a strong pillar in addressing the complexity of targeting more than 250 cancer diseases. Via Krishan Maggon
Via Krishan Maggon
Krishan Maggon 's curator insight,
April 16, 2015 7:12 AM
BTG-ADCs are stable in vivo, without DAR variation over 2 weeks, with a lower clearance compared to ADCETRIS®. In addition, quantitative time-dependent in vivo biodistribution studies show higher tumor uptake than ADCETRIS® and exhibit lower non-specific liver and spleen uptakes, resulting in higher (>3 fold) maximum tolerated dose and therefore an improved therapeutic index. |
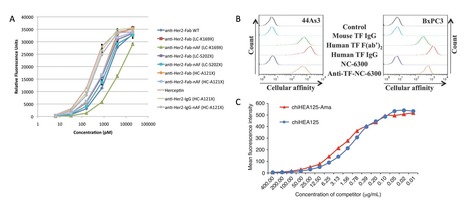
Creative biolabs offers a comprehensive analytical platform with advanced laboratory equipment to measure the customized ADC binding affinity towards the target antigen and Fc receptors.

Looking for a manufacturing partner for your antibody drug conjugates (ADCs)? Novasep has been a leading contract service provider in the ADC arena for more than 10 years....
The US FDA has cleared Seattle Genetics to restart trials of its blood cancer candidate vadastuximab talirine that were halted last year after patient deaths.
From
adcreview
A comprehensive guide to companies developing and researching antibody drug conjugates.
Via Krishan Maggon

Pharmaceutical event - Join World ADC Berlin to keep pace with the dramatic strides being made with antibody drug conjugate programs, across pre-clinical and clinical development..

In this interview we discuss a new trial involving the antibody-drug conjugate IMMU-132 as well as the future of chemotherapy in the treatment of lung cancer.

Antibody drug conjugates (ADCs) are promising therapies currently in development for oncology with unique and challenging regulatory and scientific considerations. While there are currently no regulatory guidelines specific for the nonclinical development of ADCs, there are harmonized international guidelines (e.g., ICHS6(R1), ICHM3(R2), ICHS9) that apply to ADCs and provide a framework for their complex development with issues that apply to both small and large molecules. The regulatory and scientific perspectives on ADCs are evolving due to both the advances in ADC technology and a better understanding of the safety and efficacy of ADCs in clinical development. This paper introduces the key scientific and regulatory aspects of the nonclinical development of ADCs, discusses important regulatory and scientific issues in the nonclinical to clinical dose translation of ADCs, and introduces new concepts in the areas of pharmacokinetic/pharmacodynamic (PK/PD) modeling and simulation. Via Dominique Blanchard
PLANTATION, Fla., June 9, 2015 /PRNewswire/ -- Goodwin Biotechnology, Inc., a biological Contract Development...

Abstract Via Krishan Maggon
Krishan Maggon 's curator insight,
May 24, 2015 1:57 AM
Pharmaceutical ResearchMay 2015Date: 19 May 2015Antibody-Drug Conjugates: Design, Formulation and Physicochemical StabilitySatish K. Singh, Donna L. Luisi, Roger H. Pak
From
www
Abstract Via Krishan Maggon
Krishan Maggon 's curator insight,
May 10, 2015 1:55 PM
PNAS Emanuela Romano, doi: 10.1073/pnas.1417320112Ipilimumab-dependent cell-mediated cytotoxicity of regulatory T cells ex vivo by nonclassical monocytes in melanoma patientsEmanuela Romanoa,b,c,1, Monika Kusio-Kobialkab, Periklis G. Foukasc,d, Petra Baumgaertnerc,Christiane Meyerc, Pierluigi Ballabenie, Olivier Michielina,c, Benjamin Weidef, Pedro Romeroc, andDaniel E. Speiserc Author Affiliations Edited by Ira Mellman, Genentech, Inc., South San Francisco, CA, and approved March 30, 2015 (received for review September 9, 2014)
Krishan Maggon 's curator insight,
May 12, 2015 3:05 AM
PNAS Emanuela Romano, doi: 10.1073/pnas.1417320112
Ipilimumab-dependent cell-mediated cytotoxicity of regulatory T cells ex vivo by nonclassical monocytes in melanoma patientsEmanuela Romanoa,b,c,1, Monika Kusio-Kobialkab, Periklis G. Foukasc,d, Petra Baumgaertnerc,Christiane Meyerc, Pierluigi Ballabenie, Olivier Michielina,c, Benjamin Weidef, Pedro Romeroc, andDaniel E. Speiserc Author Affiliations Edited by Ira Mellman, Genentech, Inc., South San Francisco, CA, and approved March 30, 2015 (received for review September 9, 2014)
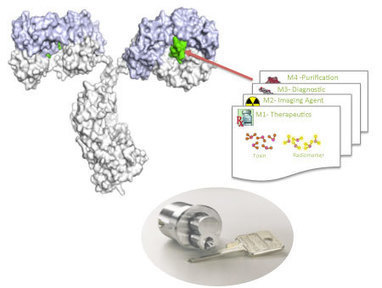
Via Krishan Maggon
Krishan Maggon 's curator insight,
April 21, 2015 7:56 AM
Meditope Biosciences' SnAP - Site-specific, novel Antibody Platform—technology is carving a new space in antibody design by turning antibodies into a "Lego-like" piece that can be combined with other pieces to expand functionality by easily 'snapping' anything imaginable on to (or off of) an antibody.

Clin Cancer Res. 2015 Apr 10. pii: clincanres.0156.2015. [Epub ahead of print]
|







