 Your new post is loading...
Your new post is loading...

|
Scooped by
NatProdChem
|
The tropics contain the overwhelming majority of Earth’s biodiversity: their terrestrial, freshwater and marine ecosystems hold more than three-quarters of all species, including almost all shallow-water corals and over 90% of terrestrial birds. However, tropical ecosystems are also subject to pervasive and interacting stressors, such as deforestation, overfishing and climate change, and they are set within a socio-economic context that includes growing pressure from an increasingly globalized world, larger and more affluent tropical populations, and weak governance and response capacities. Concerted local, national and international actions are urgently required to prevent a collapse of tropical biodiversity. The future of hyperdiverse tropical ecosystems
Jos Barlow, Filipe França, Toby A. Gardner, Christina C. Hicks, Gareth D. Lennox, Erika Berenguer, Leandro Castello, Evan P. Economo, Joice Ferreira, Benoit Guénard, Cecília Gontijo Leal, Victoria Isaac, Alexander C. Lees, Catherine L. Parr, Shaun K. Wilson, Paul J. Young & Nicholas A. J. Graham
Nature volume 559, pages517–526 (2018)
|
|
Scooped by
NatProdChem
|
Highlights- •
Metabolomics and pharmacognosy are naturally connected and cross-fertilizing. - •
Contextualization of data is needed to potentiate translational applications. - •
Data-intensive metabolomics methods unveil the need for enhanced data practices. - •
Establishing an ecosystem of open databases will nurture pharmacognosy research.
|
|
Scooped by
NatProdChem
|
|
|
Scooped by
NatProdChem
|
Guided by a “chemistry first” approach using molecular networking, eight new bright‐blue colored natural compounds, namely dactylocyanines A–H (3–10), were isolated from the Polynesian marine spong
|
|
Scooped by
NatProdChem
|
Highlights - •
LC-HRMS provides detailed, high-quality data on natural extracts metabolomes. - •
Size of spectral libraries in NPs research is limited but may be increased in silico. - •
Efficient dereplication requires a scoring combining orthogonal information. - •
Mining large datasets leads to higher confidence in the annotation of each NP. - •
Annotation of unreported metabolites represents the next challenge in NPs research.
- http://dx.doi.org/10.1016/j.cbpa.2016.12.022
Volume 36, February 2017, Pages 40–49 Omics
|
|
Scooped by
NatProdChem
|
Myxobacteria are a rich source for structurally diverse secondary metabolites with intriguing biological activities. Here we report on new natural products that were isolated from myxobacteria in the period of 2011 to July 2016. Some examples of recent advances on modes-of-action are also summarised along with a more detailed overview on five compound classes currently assessed in preclinical studies. Nat. Prod. Rep., 2017, Advance Article DOI: 10.1039/C6NP00106H
|
|
Scooped by
NatProdChem
|
Over the past few years, as the use of mass spectrometry (MS) has increased, multiple spectral libraries, databases and software frameworks have been created to enable sharing and searching of MS data. However, finding all the spectra that correspond to a specific compound across different databases continues to…
|
|
Scooped by
NatProdChem
|
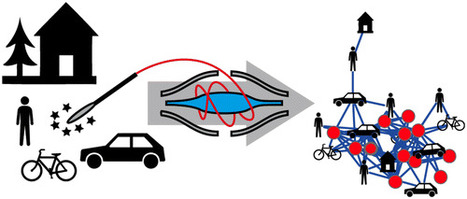
The cars we drive, the homes we live in, the restaurants we visit, and the laboratories and offices we work in are all a part of the modern human habitat. Remarkably, little is known about the diversity of chemicals present in these environments and to what degree molecules from our bodies influence the built environment that surrounds us and vice versa. We therefore set out to visualize the chemical diversity of five built human habitats together with their occupants, to provide a snapshot of the various molecules to which humans are exposed on a daily basis. The molecular inventory was obtained through untargeted liquid chromatography–tandem mass spectrometry (LC–MS/MS) analysis of samples from each human habitat and from the people that occupy those habitats. Mapping MS-derived data onto 3D models of the environments showed that frequently touched surfaces, such as handles (e.g., door, bicycle), resemble the molecular fingerprint of the human skin more closely than other surfaces that are less frequently in direct contact with humans (e.g., wall, bicycle frame). Approximately 50% of the MS/MS spectra detected were shared between people and the environment. Personal care products, plasticizers, cleaning supplies, food, food additives, and even medications that were found to be a part of the human habitat. The annotations indicate that significant transfer of chemicals takes place between us and our built environment. The workflows applied here will lay the foundation for future studies of molecular distributions in medical, forensic, architectural, space exploration, and environmental applications. Daniel Petras†‡, Louis-Félix Nothias†‡, Robert A. Quinn†‡, Theodore Alexandrov‡$▽, Nuno Bandeira‡●, Amina Bouslimani‡, Gabriel Castro-Falcón∥, Liangyu Chen†, Tam Dang‡⊗, Dimitrios J. Floros□, Vivian Hook‡, Neha Garg¶, Nicole Hoffner⊥, Yike Jiang△, Clifford A. Kapono§, Irina Koester∥, Rob Knight●■▲, Christopher A. Leber∥, Tie-Jun Ling¶&, Tal Luzzatto-Knaan¶, Laura-Isobel McCall‡, Aaron P. McGrath‡, Michael J. Meehan¶, Jonathan K. Merritt⊥, Robert H. Mills×, Jamie Morton●, Sonia Podvin‡, Ivan Protsyuk$, Trevor Purdy∥, Kendall Satterfield×%, Stephen Searles#×, Sahil Shah⊥+, Sarah Shires‡×, Dana Steffen×, Margot White∥, Jelena Todoric%, Robert Tuttle∥, Aneta Wojnicz¶, Valerie Sapp×, Fernando Vargas△, Jin Yang‡, Chao Zhang○□, and Pieter C. Dorrestein*†‡▲ Anal. Chem., Article ASAP DOI: 10.1021/acs.analchem.6b03456
|
|
Scooped by
NatProdChem
|
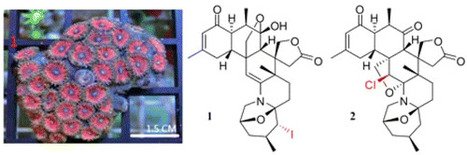
Zoanthus kuroshio is a colorful zoanthid with a fluorescent pink oral disc and brown tentacles, which dominates certain parts of the Taiwanese and Japanese coasts. This sea anemone is a rich source of biologically active alkaloids. In the current investigation, two novel halogenated zoanthamines [5α-iodozoanthenamine (1) and 11β-chloro-11-deoxykuroshine A (2)], along with four new zoanthamines [18-epi-kuroshine A (3), 7α-hydroxykuroshine E (4), 5α-methoxykuroshine E (5), and 18-epi-kuroshine E (6)], and six known compounds were isolated from Z. kuroshio. Compounds 1 and 2 are the first examples of halogenated zoanthamine-type alkaloids isolated from nature. Compounds 3 and 6 are the first zoanthamine stereoisomers with a cis-junction of the A/B rings. All isolated compounds were evaluated for their anti-inflammatory activities by measuring their effects on superoxide anion generation and elastase release by human neutrophils in response to fMLP. J. Nat. Prod., Article ASAP DOI: 10.1021/acs.jnatprod.6b00625
|
|
Scooped by
NatProdChem
|
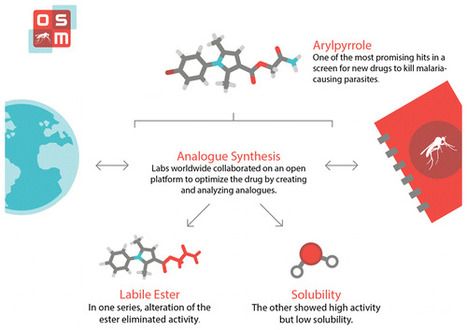
The development of new antimalarial compounds remains a pivotal part of the strategy for malaria elimination. Recent large-scale phenotypic screens have provided a wealth of potential starting points for hit-to-lead campaigns. One such public set is explored, employing an open source research mechanism in which all data and ideas were shared in real time, anyone was able to participate, and patents were not sought. One chemical subseries was found to exhibit oral activity but contained a labile ester that could not be replaced without loss of activity, and the original hit exhibited remarkable sensitivity to minor structural change. A second subseries displayed high potency, including activity within gametocyte and liver stage assays, but at the cost of low solubility. As an open source research project, unexplored avenues are clearly identified and may be explored further by the community; new findings may be cumulatively added to the present work. Alice E. Williamson†, Paul M. Ylioja†, Murray N. Robertson†, Yevgeniya Antonova-Koch§, Vicky Avery∥, Jonathan B. Baell⊥, Harikrishna Batchu#, Sanjay Batra#, Jeremy N. Burrows¶, Soumya Bhattacharyya#, Felix Calderonα, Susan A. Charman⊥, Julie Clarkβ, Benigno Crespoα, Matin Dean†, Stefan L. Debbertγ, Michael Delvesδ, Adelaide S. M. Dennisϵ, Frederik Derooseζ, Sandra Duffy∥, Sabine Fletcher∥, Guri Giaeverη, Irene Hallyburtonθ, Francisco-Javier Gamoα, Marinella Gebbiaη, R. Kiplin Guyβ, Zoe Hungerford†, Kiaran Kirkϵ, Maria J. Lafuente-Monasterioα, Anna Leeη, Stephan Meister§, Corey Nislowη, John P. Overingtonι, George Papadatosι, Luc Patinyκ, James Phamλ, Stuart A. Ralphλ, Andrea Rueckerδ, Eileen Ryan⊥, Christopher Southanμ, Kumkum Srivastava#, Chris Swainν, Matthew J. Tarnowski†, Patrick Thomsonξ, Peter Turner†, Iain M. Wallaceι, Timothy N. C. Wells¶, Karen White⊥, Laura White†, Paul Willis¶, Elizabeth A. Winzeler§, Sergio Wittlinπ, and Matthew H. Todd*† ACS Cent. Sci., Article ASAP DOI: 10.1021/acscentsci.6b00086 Publication Date (Web): September 14, 2016
|
|
Scooped by
NatProdChem
|
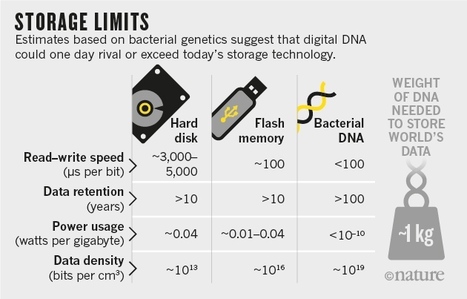
Modern archiving technology cannot keep up with the growing tsunami of bits. But nature may hold an answer to that problem already.
|
|
Scooped by
NatProdChem
|
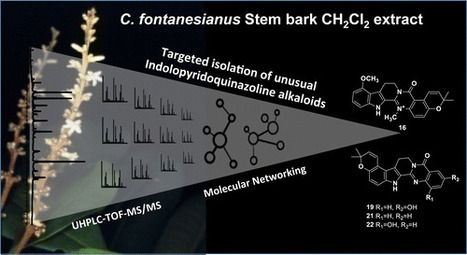
A dichloromethane-soluble fraction of the stem bark of Conchocarpus fontanesianus showed antifungal activity against Candida albicans in a bioautography assay. Off-line high-pressure liquid chromatography activity-based profiling of this extract enabled a precise localization of the compounds responsible for the antifungal activity that were isolated and identified as the known compounds flindersine (17) and 8-methoxyflindersine (18). As well as the identification of the bioactive principles, the ultra-high-pressure liquid chromatography–high-resolution mass spectrometry metabolite profiling of the dichloromethane stem bark fraction allowed the detection of more than 1000 components. Some of these could be assigned putatively to secondary metabolites previously isolated from the family Rutaceae. Generation of a molecular network based on MS2 spectra indicated the presence of indolopyridoquinazoline alkaloids and related scaffolds. Efficient targeted isolation of these compounds was performed by geometric transfer of the analytical high-pressure liquid chromatography profiling conditions to preparative medium-pressure liquid chromatography. This yielded six new indolopyridoquinazoline alkaloids (5, 16, 19–22) that were assigned structurally. The medium-pressure liquid chromatography separations afforded additionally 16 other compounds. This work has demonstrated the usefulness of molecular networks to target the isolation of new natural products and the value of this approach for dereplication. A detailed analysis of the constituents of the stem bark of C. fontanesianus was conducted. J. Nat. Prod., Article ASAP DOI: 10.1021/acs.jnatprod.6b00379
|
|
Scooped by
NatProdChem
|
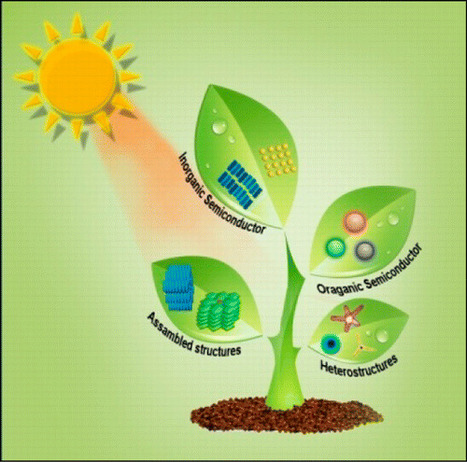
Recent advances and the current status of challenging light-harvesting nanomaterials, such as semiconducting quantum dots (QDs), metal nanoparticles, semiconductor–metal heterostructures, π-conjugated semiconductor nanoparticles, organic–inorganic heterostructures, and porphyrin-based nanostructures, have been highlighted in this review. The significance of size-, shape-, and composition-dependent exciton decay dynamics and photoinduced energy transfer of QDs is addressed. A fundamental knowledge of these photophysical processes is crucial for the development of efficient light-harvesting systems, like photocatalytic and photovoltaic ones. Again, we have pointed out the impact of the metal-nanoparticle-based surface energy transfer process for developing light-harvesting systems. On the other hand, metal–semiconductor hybrid nanostructures are found to be very promising for photonic applications due to their exciton–plasmon interactions. Potential light-harvesting systems based on dye-doped π-conjugated semiconductor polymer nanoparticles and self-assembled structures of π-conjugated polymer are highlighted. We also discuss the significance of porphyrin-based nanostructures for potential light-harvesting systems. Finally, the future perspective of this research field is given. Department of Materials Science, Indian Association for the Cultivation of Science, Jadavpur, Kolkata 700032, India Chem. Rev., Article ASAP DOI: 10.1021/acs.chemrev.6b00036 Publication Date (Web): August 5, 2016
|
|
|
Scooped by
NatProdChem
|
iNaturalist.org is a social network for naturalists! Record your observations of plants and animals,
share them with friends and researchers, and learn about the natural world.
|
|
Scooped by
NatProdChem
|
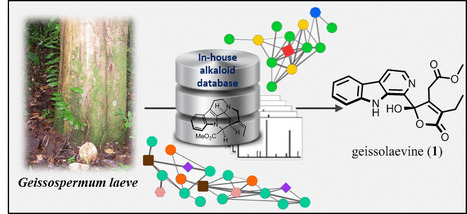
Resolving the (19R) Absolute Configuration of Lanciferine, a Monoterpene Indole Alkaloid from Alstonia boulindaensis
|
|
Scooped by
NatProdChem
|
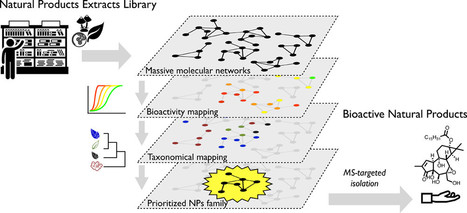
Bioactive Natural Products Prioritization Using Massive Multi-informational Molecular Networks
|
|
Scooped by
NatProdChem
|
Alexander E. Fox Ramos†, Charlotte Alcover†, Laurent Evanno†, Alexandre Maciuk†, Marc Litaudon‡ , Christophe Duplais§, Guillaume Bernadat# , Jean-François Gallard‡, Jean-Christophe Jullian†, Elisabeth Mouray⊥, Philippe Grellier⊥, Philippe M. Loiseau∥, Sébastien Pomel∥, Erwan Poupon†, Pierre Champy*†, and Mehdi A. Beniddir*† † Équipe “Pharmacognosie-Chimie des Substances Naturelles” BioCIS, Univ. Paris-Sud, CNRS, Université Paris-Saclay, 5 Rue J.-B. Clément, 92290 Châtenay-Malabry, France ‡ Institut de Chimie des Substances Naturelles, CNRS, ICSN UPR 2301, Université Paris-Saclay, 21 Avenue de la Terrasse, 91198 Gif-sur-Yvette, France § CNRS, UMR8172 EcoFoG, AgroParisTech, Cirad, INRA, Université des Antilles, Université de Guyane, 23 Avenue Pasteur, 97300 Cayenne, France # Équipe “Molécules Fluorées et Chimie Médicinale” BioCIS, Univ. Paris-Sud, CNRS, Université Paris-Saclay, 5 rue J.-B. Clément, 92290 Châtenay-Malabry, France ⊥ Unité Molécules de Communication et Adaptation des Microorganismes (MCAM, UMR 7245), Muséum National d’Histoire Naturelle, CNRS, Sorbonne Universités, CP52, 57 Rue Cuvier, 75005 Paris, France ∥ Équipe “Chimiothérapie Antiparasitaire” BioCIS, Univ. Paris-Sud, CNRS, Université Paris-Saclay, 5 Rue J.-B. Clément, 92290 Châtenay-Malabry, France J. Nat. Prod., Article ASAP DOI: 10.1021/acs.jnatprod.6b01013
|
|
Scooped by
NatProdChem
|
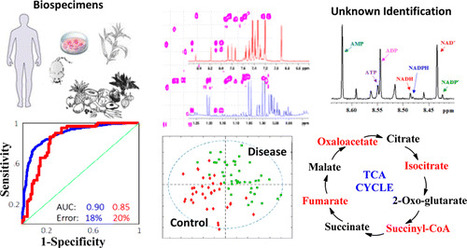
†Northwest Metabolomics Research Center, Department of Anesthesiology and Pain Medicine and ‡Department of Chemistry, University of Washington, Seattle, Washington 98109, United States, § Fred Hutchinson Cancer Research Center, Seattle, Washington 98109, United States Anal. Chem., Article ASAP DOI: 10.1021/acs.analchem.6b04420 Click here to edit the content
|
|
Scooped by
NatProdChem
|
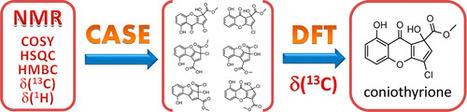
Structure elucidation of complex natural products and new organic compounds remains a challenging problem. To support this endeavor, CASE (computer-assisted structure elucidation) expert systems were developed. These systems are capable of generating a set of all possible structures consistent with an ensemble of 2D NMR data followed by selection of the most probable structure on the basis of empirical NMR chemical shift prediction. However, in some cases, empirical chemical shift prediction is incapable of distinguishing the correct structure. Herein, we demonstrate for the first time that the combination of CASE and density functional theory (DFT) methods for NMR chemical shift prediction allows the determination of the correct structure even in difficult situations. An expert system, ACD/Structure Elucidator, was used for the CASE analysis. This approach has been tested on three challenging natural products: aquatolide, coniothyrione, and chiral epoxyroussoenone. This work has demonstrated that the proposed synergistic approach is an unbiased, reliable, and very efficient structure verification and de novo structure elucidation method that can be applied to difficult structural problems when other experimental methods would be difficult or impossible to use. J. Nat. Prod., Article ASAP DOI: 10.1021/acs.jnatprod.6b00799
|
|
Scooped by
NatProdChem
|
Click hFew secondary metabolites have been reported from mammalian microbiome bacteria despite the large numbers of diverse taxa that inhabit warm-blooded higher vertebrates. As a means to investigate natural products from these microorganisms, an opportunistic sampling protocol was developed, which focused on exploring bacteria isolated from roadkill mammals. This initiative was made possible through the establishment of a newly created discovery pipeline, which couples laser ablation electrospray ionization mass spectrometry (LAESIMS) with bioassay testing, to target biologically active metabolites from microbiome-associated bacteria. To illustrate this process, this report focuses on samples obtained from the ear of a roadkill opossum (Dideiphis virginiana) as the source of two bacterial isolates (Pseudomonas sp. and Serratia sp.) that produced several new and known cyclic lipodepsipeptides (viscosin and serrawettins, respectively). These natural products inhibited biofilm formation by the human pathogenic yeast Candida albicans at concentrations well below those required to inhibit yeast viability. Phylogenetic analysis of 16S rRNA gene sequence libraries revealed the presence of diverse microbial communities associated with different sites throughout the opossum carcass. A putative biosynthetic pathway responsible for the production of the new serrawettin analogues was identified by sequencing the genome of the Serratia sp. isolate. This study provides a functional roadmap to carrying out the systematic investigation of the genomic, microbiological, and chemical parameters related to the production of natural products made by bacteria associated with non-anthropoidal mammalian microbiomes. Discoveries emerging from these studies are anticipated to provide a working framework for efforts aimed at augmenting microbiomes to deliver beneficial natural products to a host.ere to edit the content
|
|
Scooped by
NatProdChem
|
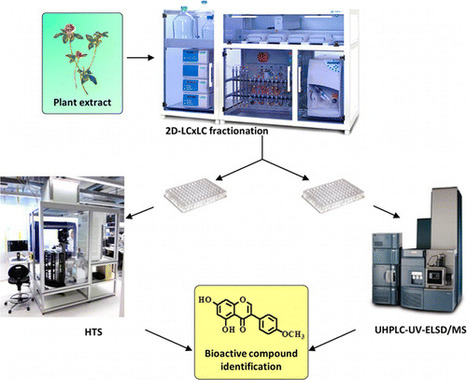
To identify natural bioactive compounds from complex mixtures such as plant extracts, efficient fractionation for biological screening is mandatory. In this context, a fully automated workflow based on two-dimensional liquid chromatography (2D-LC × LC) was developed, allowing for the production of hundreds of semipure fractions per extract. Moreover, the ELSD response was used for online sample weight estimation and automated concentration normalization for subsequent bioassays. To evaluate the efficiency of this protocol, an enzymatic assay was developed using AMP-activated protein kinase (AMPK). The activation of AMPK by nonactive extracts spiked with biochanin A, a known AMPK activator, was enhanced greatly when the fractionation workflow was applied compared to screening crude spiked extracts. The performance of the workflow was further evaluated on a red clover (Trifolium pratense) extract, which is a natural source of biochanin A. In this case, while the crude extract or 1D chromatography fractions failed to activate AMPK, semipure fractions containing biochanin A were readily localized when produced by the 2D-LC×LC-ELSD workflow. The automated fractionation methodology presented demonstrated high efficiency for the detection of bioactive compounds at low abundance in plant extracts for high-throughput screening. This procedure can be used routinely to populate natural product libraries for biological screening. Paul Coulerie†‡, Yann Ratinaud†, Sofia Moco†, Loraine Merminod†, Martine Naranjo Pinta†, Julien Boccard‡, Laurent Bultot†, Maria Deak†, Kei Sakamoto†, Emerson Ferreira Queiroz‡, Jean-Luc Wolfender‡, and Denis Barron*† DOI: 10.1021/acs.jnatprod.6b00628 Publication Date (Web): October 28, 2016
|
|
Scooped by
NatProdChem
|
Centrifugal partition chromatography (CPC) and all countercurrent separation apparatus provide chemists with efficient ways to work with complex matrixes, especially in the domain of natural products. However, despite the great advances provided by these techniques, more efficient ways of analyzing the output flow would bring further enhancement. This study describe a hyphenated approach made by coupling NMR with CPC through a hybrid-indirect coupling made possible by using a solid phase extraction (SPE) apparatus intended for high-pressure liquid chromatography (HPLC)-NMR hyphenation. Some hardware changes were needed to adapt the incompatible flow-rates and a reverse-engineering approach that led to the specific software required to control the apparatus. 1D 1HNMR and 1H–1H correlation spectroscopy (COSY) spectra were acquired in reasonable time without the need for any solvent-suppression method thanks to the SPE nitrogen drying step. The reduced usage of expensive deuterated solvents from several hundreds of milliliters to the milliliter order is the major improvement of this approach compared to the previously published ones. Anal. Chem., Article ASAP DOI: 10.1021/acs.analchem.6b01429
|
|
Scooped by
NatProdChem
|
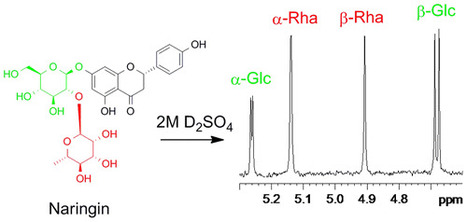
The sugar subunits of natural glycosides can be conveniently determined by acid hydrolysis and 1H NMR spectroscopy without isolation or derivatization. The chemical shifts, coupling constants, and integral ratios of the anomeric signals allow each monosaccharide to be identified and its molar ratio to other monosaccharides to be quantified. The NMR data for the anomeric signals of 28 monosaccharides and three disaccharides are reported. Application of the method is demonstrated with the flavonoid glycoside naringin (1), the aminoglycoside antibiotics kanamycin (2) and tobramycin (3), and the saponin digitonin (4). J. Nat. Prod., Article ASAP DOI: 10.1021/acs.jnatprod.6b00180
|
|
Scooped by
NatProdChem
|
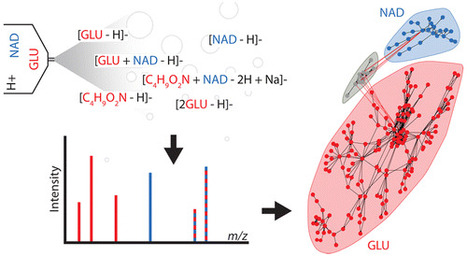
Analysis of a single analyte by mass spectrometry can result in the detection of more than 100 degenerate peaks. These degenerate peaks complicate spectral interpretation and are challenging to annotate. In mass spectrometry-based metabolomics, this degeneracy leads to inflated false discovery rates, data sets containing an order of magnitude more features than analytes, and an inefficient use of resources during data analysis. Although software has been introduced to annotate spectral degeneracy, current approaches are unable to represent several important classes of peak relationships. These include heterodimers and higher complex adducts, distal fragments, relationships between peaks in different polarities, and complex adducts between features and background peaks. Here we outline sources of peak degeneracy in mass spectra that are not annotated by current approaches and introduce a software package called mz.unity to detect these relationships in accurate mass data. Using mz.unity, we find that data sets contain many more complex relationships than we anticipated. Examples include the adduct of glutamate and nicotinamide adenine dinucleotide (NAD), fragments of NAD detected in the same or opposite polarities, and the adduct of glutamate and a background peak. Further, the complex relationships we identify show that several assumptions commonly made when interpreting mass spectral degeneracy do not hold in general. These contributions provide new tools and insight to aid in the annotation of complex spectral relationships and provide a foundation for improved data set identification. Mz.unity is an R package and is freely available at https://github.com/nathaniel-mahieu/mz.unity as well as our laboratory Web site http://pattilab.wustl.edu/software/. Anal. Chem., Article ASAP DOI: 10.1021/acs.analchem.6b01702
|
|
Scooped by
NatProdChem
|
The Research Ideas and Outcomes (RIO) journal publishes all outputs of the research cycle, including: project proposals, data, methods, workflows, software, project reports and research articles together on a single collaborative platform, with the most transparent, open and public peer-review process. Our scope encompasses all areas of academic research, including science, technology, humanities and the social sciences.
|



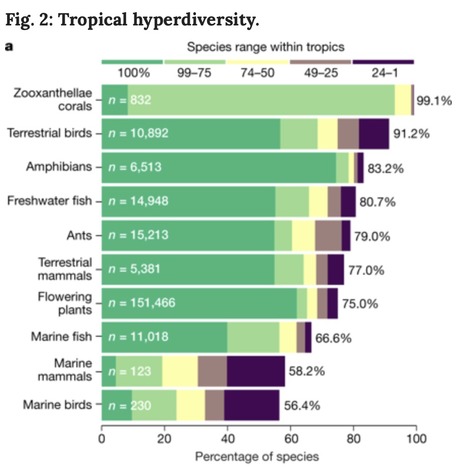













The future of hyperdiverse tropical ecosystems ... our treasures !