 Your new post is loading...

|
Scooped by
?
Today, 12:50 PM
|
Phosphate is often a limiting resource, directly affecting the availability of key biomolecules such as nucleotides. To cope with phosphate scarcity, bacteria have evolved enzymes that utilize alternative phosphorus compounds, including phosphite (Pt). Although a few enzymes oxidize Pt to produce phosphate, the enzymes responsible for Pt oxidation in many environmental bacteria remain unidentified, and the role of microbial Pt oxidation in the global phosphorus cycle is not yet fully understood. In this study, we performed bioinformatic analyses of three Pt-oxidizing enzymes: the native Pt oxidase, phosphite dehydrogenase (PtxD), and two promiscuous Pt oxidases, alkaline phosphatase (PhoA) and carbon–phosphorus lyase. Among these, PhoA was found to be widely distributed across bacteria since the early stages of their evolution. In contrast, PtxD emerged later in a limited number of bacterial lineages that had lost PhoA. Our biochemical characterizations revealed that most extant and reconstructed ancestral PhoAs tested exhibited Pt oxidation activity. Moreover, disruption of active-site residues diminished Pt oxidase activity in PhoA, while only partially affecting its native function. This promiscuous function of PhoA reveals an overlooked mechanism in bacterial phosphate metabolism and underscores the role of Pt in the cycling of bioavailable phosphorus in ecosystems.
|
|
Scooped by
?
Today, 12:13 PM
|
Signal transduction by histidine kinases (HKs) is nearly ubiquitous in bacterial species. HKs can either sense ligands directly or indirectly via a cognate solute-binding protein (SBP). The molecular basis for SBP-dependent signal reception, however, remains poorly understood in most cases. CBP and ChiS are the SBP–HK pair that activate the chitin utilization program of Vibrio cholerae. Here, we elucidate the molecular basis for allosteric regulation of CBP–ChiS by generating structural models of this complex in the unliganded and liganded states, which we support with extensive genetic, biochemical, and cell biological analysis. Our results reveal that ligand-binding induces a large conformational interface switch that is distinct from previously described SBP–HKs. Structural modeling suggests that similar interface switches may also regulate other uncharacterized SBP–HKs. Together, these results extend our understanding of signal transduction in bacterial species and highlight an approach for uncovering the molecular basis of allostery in protein complexes.
|
|
Scooped by
?
Today, 11:12 AM
|
Cell type annotation is an essential step in single-cell RNA-sequencing analysis, and numerous annotation methods are available. Most require a combination of computational and domain-specific expertise, and they frequently yield inconsistent results that can be challenging to interpret. Large language models have the potential to expand accessibility while reducing manual input and improving accuracy, but existing approaches suffer from hyperconfidence, hallucinations, and lack of reasoning. To address these limitations, we developed CASSIA for automated, accurate, and interpretable cell annotation of single-cell RNA-sequencing data. As demonstrated in analyses of 970 cell types, CASSIA improves annotation accuracy in benchmark datasets as well as complex and rare cell populations, and also provides users with reasoning and quality assessment to ensure interpretability, guard against hallucinations, and calibrate confidence. Assigning cell types in single-cell RNA-seq is essential yet challenging, as it requires expertise, time, and is often subjective. Here, the authors present CASSIA, a multi-agent AI system that provides automated, interpretable, and quality-controlled annotations with high accuracy.
|
|
Scooped by
?
Today, 11:03 AM
|
DNA language models offer a new paradigm for sequence design, yet their ability to generate functional genomic sequences remains underexplored. Plasmids act as a good testbed for evaluating DNA language model generation potential due to their simplicity and ease of construction. Here, we develop an end-to-end pipeline for generative design of Escherichia coli plasmid backbones, from large-scale data curation through fine-tuning, sampling, bioinformatic assessment, and candidate selection. A curated plasmid library was assembled from PlasmidScope and Addgene, and PlasmidGPT, a GPT-2-style DNA model, was fine-tuned on these corpora using circular-aware batching and random crops. Generations (1,000 per model) were produced under two prompting strategies: a minimal ATG seed to expose default tendencies, and a GFP cassette to enforce functional context. From 1000 generated synthetic plasmids, 16 candidates survived strict filtering and these were prioritised for wet-lab validation. Three shortlisted plasmids were synthesised and found to be functional, supporting growth, antibiotic resistance, and GFP expression in E. coli. These represent, to our knowledge, the first full AI-generated plasmids to be synthesised and validated in vivo. This work demonstrates that curated fine-tuning and prompt-aware generation enable DNA language models to progress from raw sequence sampling to experimentally testable plasmid designs. The approach offers a foundation for extending DNA design optimisation beyond E. coli, toward broader applications across engineering biology.
|
|
Scooped by
?
Today, 12:00 AM
|
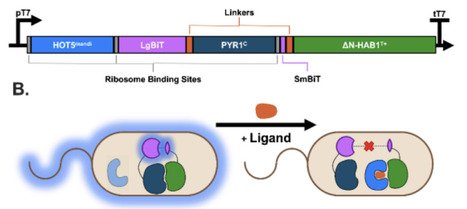
Rational control over diverse, ligand-responsive output networks is a foundational challenge in synthetic biology, particularly for systems based on post-translational signaling. Here, we present the design and engineering of minimal, modular protein architectures for chemically responsive molecular inverters and digital switches. Our inverter design modifies the plant-derived PYR1-HAB1 chemically inducible dimerization module by incorporating a constitutive activator, which is then competitively displaced by the ligand-bound PYR1 receptor, converting the native 'dimerization-on' mechanism into a 'signal-off' inverter. We establish key design features and demonstrate predictable tuning of the inverter's transfer function, including its maximum output, half-maximal inhibitory concentration, and minimum output, solely by adjusting protein stoichiometry. The architecture is modular, enabling plug-and-play response to diverse, user-defined drug-like small molecules. We show that an inverter biosensor for an environmental contaminant functions in engineered living cells with a low nanomolar sensitivity. Additionally, we convert the PYR1-HAB1 sensor into a digital switch by adding an engineered molecular titrant to the system. Overall, this work provides a generalizable, minimal, and tunable protein scaffold for programming complex, post-translational signaling logic, significantly expanding the toolkit for sophisticated biological circuit design.
|
|
Scooped by
?
December 6, 11:27 PM
|
Sporosarcina pasteurii is the most widely studied bacterium for microbially-induced calcium carbonate precipitation (MICP), a process of intense interest for materials and construction applications. Despite two decades of investigation, S. pasteurii has remained genetically intractable, limiting our mechanistic understanding of biomineralization pathways and constraining efforts to engineer scalable solutions. Here, we present the first genetic toolkit for S. pasteurii, including a stable replicating plasmid, a conjugation-based DNA delivery protocol, engineered inducible promoters, and methods for genome modification. Using homologous recombination, we precisely deleted 5.7 kb of the genome spanning two operons encoding urease activity and demonstrated complete loss of biocementation. We also screened a library of engineered transposon constructs for activity in S. pasteurii and generated a genome-wide mutant library with >15,000 unique insertion sites. Using this library, we identified putative genes affecting ureolytic growth, revealing previously inaccessible aspects of S. pasteurii genetics. This work establishes S. pasteurii as a genetically tractable platform for rational engineering of MICP and constitutes the first genetic modification capability within the Sporosarcina genus.
|
|
Scooped by
?
December 6, 11:06 PM
|
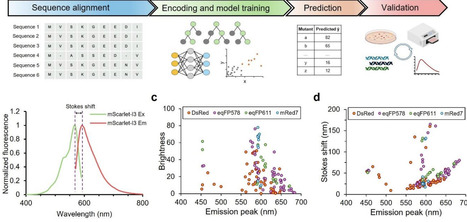
Fluorescent proteins (FPs) are widely used reporters for visualizing cellular structures and processes. Traditional wet-lab strategies for FP engineering (rational design and directed evolution) have enabled substantial improvements in photophysical performance but are limited by their requirement for deep expert knowledge or labor-intensive screening. AI-driven approaches have recently gained traction for engineering variants of green FPs, yet applications to red fluorescent proteins (RFPs) remain scarce. Here, we applied machine learning models to an RFP sequence-function dataset and trained these models to predict functional single-mutation variants of the state-of-the-art RFP mScarlet-I3. Guided by model predictions, we identified variants exhibiting red-shifted emission peaks, large Stokes shifts, or brightness comparable to the parental protein. Our findings show that even lightweight, data-efficient models can extract actionable design principles for improving RFPs. This work demonstrates the feasibility of AI-guided design for RFPs and provides a reliable benchmark for future development of more powerful AI-driven strategies for FP engineering.
|
|
Scooped by
?
December 6, 10:53 PM
|
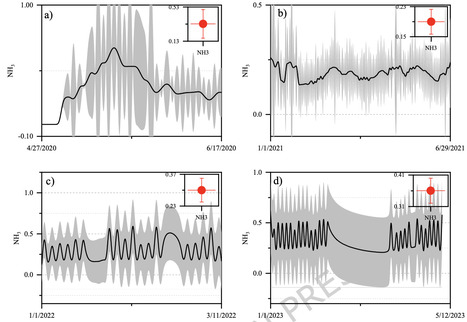
This study presents a LoRaWAN-based IoT system developed for real-time monitoring of ammonia (NH₃) emissions in cereal crop fields. Sustainable agriculture increasingly demands on-farm greenhouse gas (GHG) tracking linked to environmental variables. IoT offers efficient real-time monitoring of soil NH₃ emissions and associated factors. Our research introduces a unique Field Monitoring Laboratory: a LoRaWAN-connected IoT system integrating soil, crop, and microclimate sensors to observe NH₃⁺, air temperature, rainfall, humidity, soil temperature, and moisture content. The system comprises a field lab, data server, and custom dashboard with analytics capabilities. NH₃ fluxes were measured in autumn-sown cereals across three growing seasons (2020–2023). Tukey’s Kramer test revealed significant (p < 0.05, p < 0.001) differences in NH₃ emissions and environmental variables between years. Highest NH₃ emissions (1.94 ppm in 2020, 1.71 ppm in 2021) coincided with elevated air (25–31 °C) and soil (21–23 °C) temperatures, and higher mean and peak rainfall (0.40–0.48 mm average; max 9–31.6 mm). Principal Component Analysis showed 65.8% variance explained by PC1 and PC2, with high loadings from temperature and soil moisture. Spearman’s correlation indicated moderate positive associations (r = 0.38–0.4, p < 0.05) of NH₃ with soil moisture at 20 cm and 40 cm of soil depth, and a weak negative correlation (r = -0.16 and − 0.17) with soil temperature at 20 cm and 40 cm. The study underscores the potential of IoT technology using calibrated gas sensors and LoRaWAN for real-time NH₃ and environmental monitoring, enabling informed decision-making in smart agriculture.
|
|
Scooped by
?
December 6, 10:41 PM
|
The methylotrophic yeast Pichia pastoris (also known as Komagataella phaffii) is a prominent platform for recombinant protein production, offering benefits such as thermo- and osmotolerance, high-density growth, and efficient protein secretion. Its ability to metabolize methanol, an increasingly available carbon source, enhances its cost-effectiveness and sustainability for industrial use. As a eukaryotic host, P. pastoris ensures proper protein folding and post-translational modifications (PTMs), including glycosylation, which is essential for correct folding and endoplasmic reticulum (ER) quality control. While ER-transferred glycans are critical for maturation, additional modification in the Golgi apparatus can yield larger glycans whose impact on stability, solubility, and bioactivity may be either beneficial or undesirable, depending on the application of the heterologous protein. The impact of induction conditions on glycosylation of proteins secreted by P. pastoris SuperMan5 was examined, using the DS-1 (G2P[4]) and WA (G1P[8]) VP8* rotavirus capsid proteins as a model. An ELISA-based screening system was employed for clone selection and media optimization, with results showing easy integration into automated workflows. Methanol concentration was found to impact both N- and O-linked glycosylation complexity, shaping the glycosylation profile of the target protein as well as the P. pastoris secretome. This study underscores the importance of optimizing cultivation conditions to enhance protein yield, refine glycosylation, and minimise impurities, all of which are crucial for large-scale production and efficient downstream processing. It also suggests a method for easy modulation of glycosylation depending on the target application and the desired level of glycosylation.
|
|
Scooped by
?
December 6, 3:52 PM
|
Protein glycosylation is a very common post-translational modification PTM seen in all branches of biology. The functional roles for protein glycosylation are many and varied, essential in eukaryotes but seemingly dispensable in bacteria. One group of bacteria where protein glycosylation has been looked at for at least 50 years are the actinobacteria, a large and diverse group of bacteria which include well know pathogens like Mycobacteria tuberculosis, Corynebacterium diphtheriae, and well know species important in biotechnology like Streptomyces lividans and Corynebacterium glutamicum. Actinobacterial protein glycosylation is a form of protein O-mannosylation which is found widely in eukaryotes from yeast on up the evolutionary ladder but is much less understood at the functional level. Very few direct roles for protein O-mannosylation have been described in the literature. This review examines newer findings from the actinobacterial world which with the help of glycoprotein models suggests how the glycans might play a role in actinobacterial biology.
|
|
Scooped by
?
December 6, 3:26 PM
|
Ammonia-oxidizing archaea (AOA) are among the most abundant microorganisms in the ocean and play a critical role in marine nitrogen cycling. Recently, urea has been shown to serve as an additional substrate for marine AOA, with substantial urea use in the ammonium-depleted open-ocean. Yet, the mechanisms that control urea use and potentially maintain high AOA abundances remain unclear. Here, we investigate urea and ammonia use by AOA in three contrasting marine environments, from coastal, ammonium-rich to open-ocean, ammonium-poor waters. Our combined results indicate that distinct substrate utilization strategies of Nitrosopumilus and Nitrosopelagicus control their environmental distribution. The more coastal AOA genus, Nitrosopumilus, primarily uses ammonium. In contrast, enhanced urea utilization in ammonium-limited waters is linked to the activity and growth of Nitrosopelagicus. Thus, the use of urea, and potentially other organic-N compounds by Nitrosopelagicus plays a major role in fueling open-ocean nitrification and sustaining primary productivity in these vast regions. Two groups of ammonia-oxidizing archaea drive marine nitrification. Stuehrenberg et al. reveal that their distribution reflects substrate use, with one relying on urea and the other on ammonia to maintain nitrification in open-ocean waters.
|
|
Scooped by
?
December 6, 3:00 PM
|
Plant protein production systems are scalable and sustainable platforms capable of meeting the growing demand for functional proteins in nutrition, pharmaceuticals, and industry. Recent advances in essential amino acid (EAA) biosynthesis, gene regulation, and subcellular targeting have enhanced protein yields and stability, but are yet to be integrated into holistic engineering approaches. Metabolic engineering can improve amino acid (AA) metabolism and energy efficiency, while genetic engineering enables finetuned, spatiotemporal expression of target proteins. Coupled with in silico tools for protein design, novel proteins with enhanced stability and functionality can be developed. Integrating these strategies would enable the fine-tuning of protein synthesis while balancing cellular energy costs, offering context-dependent opportunities to advance protein production in plant systems.
|
|
Scooped by
?
December 6, 2:51 PM
|
Reactive oxygen species (ROS) are a promising alternative bactericide. However, it is questioned that bacteria can potentially develop resistance to ROS, similar to their resistance against antibiotics and silver. Herein, it is reported that Gram-negative bacteria, including Pseudomonas aeruginosa, Escherichia coli, and Klebsiella pneumoniae, develop resistance to ROS after six repeated exposures. Notably, ROS minimum inhibitory concentration of P. aeruginosa significantly increases to 256-fold after ten passages. The resistance mechanism predominantly originates from the intensified biosynthesis of the highly reductive hydrogen sulfide (H2S) and pyoverdine (PVD) siderophores, effectively neutralizing ROS. Simultaneously, PVD transports Fe3+ from the extracellular space into the bacteria, releasing H2S bound to Fe3+ and enhancing ROS scavenging. Additionally, the enhanced outer membrane (OM) biogenesis establishes a robust OM barrier, impeding ROS penetration. The acquired resistance to ROS can be significantly reduced by incorporating additional Fe3+ into the culture medium or disrupting the H2S biosynthetic gene. These observations suggest that careful consideration is required when utilizing ROS against Gram-negative bacteria. It is anticipated that understanding this resistance mechanism can inform the development of future antimicrobial agents, particularly for Gram-negative bacteria.
|
|
|
Scooped by
?
Today, 12:46 PM
|
Bacterial small RNAs (sRNAs) derived from mRNA 3′ untranslated regions (3′ UTRs) have emerged as important regulators of gene expression, yet their evolutionary origins and functional diversification remain poorly understood compared to the protein-coding sequences of the same transcripts. In this study, we present a comparative analysis of the biogenesis and regulatory functions of UhpU, a 3′ UTR–derived sRNA from the hexose phosphate transporter gene uhpT, in Escherichia and Salmonella. We show that UhpU likely originated as a processed sRNA generated by RNase E cleavage in Enterobacteriaceae, enabling repression of genes in the hexose phosphotransferase system, thereby contributing to hexose phosphate homeostasis in coordination with its parental gene. In Escherichia, UhpU subsequently evolved the ability to repress mprA, a transcriptional repressor, via a UhpU-binding site introduced by a horizontally acquired DNA fragment that extended the mprA 5′ UTR. After divergence from the most recent common ancestor with Escherichia albertii, the lineage comprising Escherichia coli, Escherichia fergusonii, and Shigella acquired a FliA-dependent promoter within the uhpT coding region, allowing independent transcription of UhpU and establishing it as a dual-biogenesis sRNA. Together, our results outline a stepwise trajectory in which UhpU evolved from a processing-derived metabolic regulator to an sRNA with expanded regulatory connections and a lineage-specific FliA-dependent transcriptional program in Escherichia.
|
|
Scooped by
?
Today, 11:27 AM
|
Retrons are bacterial genetic retroelements encoding a reverse transcriptase (RT) and a non-coding RNA (ncRNA)-multi-copy single-stranded DNA (msDNA) hybrid. Diverse effector proteins or domains are found to associate with retrons, typically forming tripartite toxin-antitoxin systems involved in antiphage defense. Although retrons have attracted growing interest in genome editing technologies, the mechanisms underlying most retron-mediated immune systems remain poorly understood. Here, we characterize a distinct quaternary retron system, Ec78, harboring a dual-component effector complex in which the PtuA ATPase and PtuB nuclease act in concert to mediate phage clearance. The cryo-EM structure of the Ec78 complex adopts a flower-basket-like architecture, with two Ec78 retrons engaging the PtuAB effector complexes through a msDNA-insertion assembly mechanism. Shortening of msDNA in length releases the PtuAB from Ec78 retron and triggers its activation. The cryo-EM structure of the retron-unbound effector complex further reveals an arginine-lysine finger loop on the PtuB nuclease that undergoes an ordered-to-disordered transition during enzymatic activation. These findings delineate the molecular basis underlying the Ec78 system in antiviral defense and highlight the mechanistic diversity of retron systems in prokaryotic immunity. Retrons are bacterial genetic retroelements implicated in anti-phage defense. Here, the authors characterize the quaternary retron system Ec78 and elucidate the biochemical mechanisms underlying Ec78-mediated prokaryotic immunity.
|
|
Scooped by
?
Today, 11:09 AM
|
Antimicrobial host defense peptides are promising alternatives to resistance prone small molecule antibiotics. To overcome the poor physiologic stability of these therapeutic candidates it is common to prepare proteolytically resistant retro-inverso analogues, where sequence backbone direction and amino acid chirality are reversed. However, in many cases, gains in stability are offset by altered assembly propensities and reduced biologic potency. Here, we show that, contrary to the dogma for non-mycobacterial pathogens, retro-inversion of antimycobacterial host defense peptides improves their potency, specificity and host safety; in some cases by more than an order of magnitude. Biophysical assays suggest that altered mycomembrane thermodynamics, instead of improved proteolytic stability, plays a causative role in retro-inverso mediated potency gains. Additional bacteriologic assays using a lead retro-inversed candidate, MAD1-RI, demonstrate this analogue rapidly sterilizes replicating cultures of Mycobacterium tuberculosis, is effective towards drug-resistant clinical isolates of the pathogen, and synergistically enhances the activity of co-incubated antibiotics. Transcriptomic studies uncover complementary membrane destabilizing and metabolic mechanisms of antitubercular action for MAD1-RI, and in doing so identify sequence retro-inversion as a simple, but powerful, modality in the de novo design of non-natural antimycobacterial peptides. In this work, authors show that retro-inversion of host-defense peptides markedly improves their antitubercular potency and selectivity, highlighting the potential of this simple chemical modification to advance the design of novel antimycobacterial biotherapeutics.
|
|
Scooped by
?
Today, 12:26 AM
|
Adaptation occurs through the selection of beneficial mutations enhancing fitness in a specific environment. However, since environments vary across time and space, mutations that are positively selected in one environment may be less beneficial or detrimental in others. Here, we investigate the evolution of microbial robustness (i.e. a consistent fitness across many diverse environments) through the adaptive evolution of two genetically distinct Saccharomyces cerevisiae populations in fluctuating conditions, followed by fitness assays and whole-genome sequencing. Our results indicate that the haploid laboratory strain S288C achieved higher average fitness than its parental strain, particularly when evolved in fluctuating environments compared to constant environments, but did not show increased robustness. In contrast, populations of the industrial diploid strain Ethanol Red failed to achieve significant fitness improvement under both fluctuating and constant evolution regimes but became more robust. Populations that adapted to fluctuating conditions acquired mutations in genes involved with cell morphology and protein degradation. Overall, our results emphasise the importance of parental traits in shaping fitness and robustness during adaptive laboratory evolution.
|
|
Scooped by
?
December 6, 11:34 PM
|
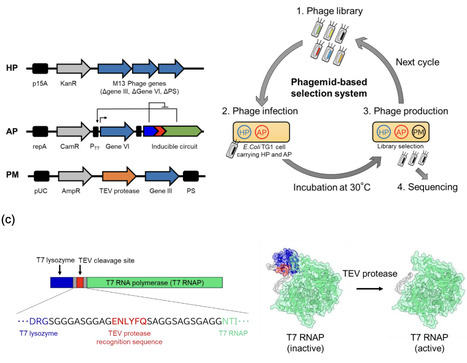
Loop engineering of enzymes remains challenging due to high flexibility and conformational complexity, posing a bottleneck for deep-learning-based design. Here, we constructed mutant libraries for three loops of TEV protease to assess combining directed evolution with deep learning. Using an M13 phagemid-based selection system, the three libraries were screened, resulting in a Loop 1 variant (HyperTEV60/L1) that significantly enhanced the Michaelis constant (Km) of the HyperTEV60 scaffold, a highly active mutant identified by ProteinMPNN. Structural modeling suggested that a single-residue deletion and substitution in Loop 1 expands the substrate binding pocket, accounting for the improved Km. Although the catalytic efficiency kcat/Km of HyperTEV60/L1 was only marginally higher than HyperTEV60, due to a kcat decrease, our results reveal that the phagemid-based selection system tended to find variants optimizing Km. This study demonstrates that combining deep-learning-based global optimization with localized directed evolution maximizes the probability of discovering distinct, high-performance enzyme variants.
|
|
Scooped by
?
December 6, 11:23 PM
|
Metagenomic taxonomic profiling is essential for characterizing microbial community composition in both environmental and clinical contexts. Existing profilers have greatly advanced community characterization; however, achieving accurate profiling across all domains of life - especially for archaea, fungi, and viruses - and for low-biomass, host-dominated samples, remains challenging. We describe Metax, a cross-domain taxonomic profiler that employs probabilistic modeling of genome coverage to distinguish true community members from artifactual signals arising from reference contamination, local genomic similarity, or reagent-derived DNA fragments. In comprehensive benchmarks across more than 500 samples, Metax demonstrated accurate species-level profiling, with consistent performance for bacteria, archaea, eukaryotes and viruses, and robustness to shallow sequencing. Applied to an oral microbiome cohort, Metax identified differentially abundant viral taxa distinguishing peri-implantitis from healthy sites, while analyses of tumor microbiome data revealed reagent-borne contaminants and potential reference misassemblies. By integrating coverage-informed statistics, Metax delivers accurate, robust, and interpretable cross-domain taxonomic profiles, maintaining stable performance across diverse sequencing depths and sample types.
|
|
Scooped by
?
December 6, 11:02 PM
|
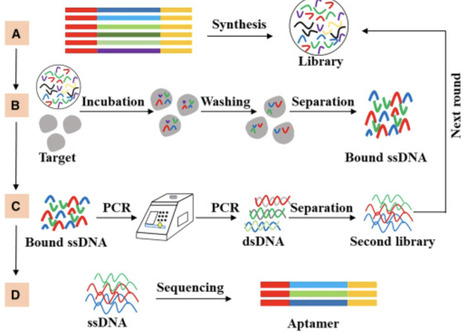
Conventional methods for plant disease detection and management often face limitations in sensitivity, specificity, and field applicability. Aptamers, with high affinity and specificity, present a promising alternative. The review begins by elucidating the molecular mechanisms and key benefits of aptamers over traditional antibodies, highlighting their robustness and versatility. Building upon their demonstrated successes in food safety, environmental monitoring, and medical diagnostics, we systematically examine the application prospects of aptamers in plant disease management. Specifically, aptamers not only served as sensitive biosensors for detecting plant hormones and pathogens but also as active agents that interfere with pathogen effectors and modulate plant immune responses. Although emerging, aptamers hold considerable potential to transform plant disease control. Future advancements will require overcoming challenges in aptamer screening and biosensor stability through integration with emerging technologies, including microfluidics, CRISPR, and artificial intelligence, paving the way for intelligent, field-deployable diagnostics for sustainable agriculture.
|
|
Scooped by
?
December 6, 10:46 PM
|
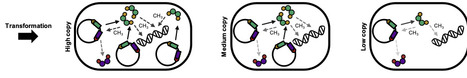
Restriction-modification (R-M) systems are one of the most widespread and, due to their often plasmid-based nature, transmittable antiphage systems bacteria have. The CfrBI R-M system studied here consists of a methyltransferase (MT) and a restriction endonuclease (RE) that are divergently expressed and share a promoter region that harbors a single CfrBI recognition site. Previously, the methylation of this site has been shown to regulate the expression of the R-M system. Here, we show that the expression dynamics of the CfrBI R-M system and its protective properties depend on the copy number of the plasmid harboring it. A higher copy number results in higher expression, but the expression on a medium-copy number plasmid interestingly conferred the highest phage resistance. After transformation of naïve cells however, expression of the RE was fastest in the high-copy plasmid background. In vivo we show that the expression strength of the MT inhibits its own expression, while enhancing RE expression. To conclude, the results indicate that for phage resistance the overall expression strength might not be the predominant factor in case of the CfrBI R-M system, while for the establishment of the system the initial expression rate of the MT seems to be the determining factor.
|
|
Scooped by
?
December 6, 10:23 PM
|
An unbiased and accurate estimation of intraspecies diversity, i.e., the extent of genetic diversity within species (or microdiversity), is crucial for clinical and environmental microbiome studies. Although it is well appreciated that sequencing depth (or coverage depth) below 10X can provide biased estimates of microdiversity, typically underestimating diversity due to the random sampling of alleles, there is a widely accepted convention that microdiversity estimates tend to be relatively stable at sequencing depth exceeding 10X. Therefore, discarding species with less than 10X or rarefying to 10-20X sequencing depth are generally used to compare microdiversity among taxa and samples. Our findings showed that these biases may persist even at depth levels above 50-200X for all popular sequencing platforms, including Illumina, PacBio, and Oxford Nanopore. The biases mostly, but not always, represent an underestimation of diversity and were attributable to the incomplete recovery of Single Nucleotide Variants (SNVs) at lower sequencing depth levels. To address this issue, we recommend using rarefaction-based approaches to standardize data at least 50X, and ideally at 200X sequencing depth, which reduces differences between observed and expected microdiversity values to less than 0.5%. Furthermore, the Average Nucleotide Identity of reads (ANIr) metric is significantly less sensitive to sequencing depth variability than nucleotide diversity (π), making it a robust alternative for estimating microdiversity at sequencing depth close or exceeding 10X, without a need to rarefying data. Therefore, the sequencing depth thresholds proposed herein provide a more standardized framework for direct comparisons of microdiversity across samples and studies.
|
|
Scooped by
?
December 6, 3:39 PM
|
Type VI secretion systems (T6SSs) are molecular machines used by bacteria to release effectors that target either host cells, competing bacteria or fungi. Regulatory mechanisms underlying antifungal T6SS activity remain unexplored. Here we show, using mouse infection with wild-type and T6SS mutant bacteria, that T6SS activity of the enteropathogen, Yersinia pseudotuberculosis (Yptb), reduces fungal prevalence in the gut microbiota and has direct activity on Candida albicans. Screening of bacterial effector mutant strains, and structural and biochemical analyses identify TfeC as an antifungal chitinase T6SS effector that can kill C. albicans. In vivo experiments confirm that TfeC expression promotes Yptb colonization and reduces C. albicans abundance. We also show that Yptb senses the fungal quorum-sensing molecule, tyrosol, through the two-component system, EnvZ–OmpR, and responds by activating T6SS4. Our findings suggest that Yptb modulates its antifungal activities by detecting changes in fungal population density cues, revealing a mechanism of fungal–bacterial interkingdom communication mediated by fungal quorum-sensing molecules. Yersinia pseudotuberculosis senses fungal tyrosol signalling through EnvZ–OmpR which triggers T6SS activation and antifungal effector release to reduce fungal competitors in the mouse gut.
|
|
Scooped by
?
December 6, 3:16 PM
|
E. coli RecBCD, a hetero-trimeric helicase and nuclease, functions in double stranded (ds) DNA break repair. RecBCD possesses ATPase motor domains within both RecB (3’ to 5’) and RecD (5’ to 3’) and a nuclease domain within RecB (RecBNuc). RecBCD binds to double stranded DNA ends and initiates DNA unwinding by first melting several DNA base pairs (bp) using only its binding free energy. The RecBNuc domain is docked ∼70 Å from the duplex DNA binding site in RecBCD-DNA structures but appears to be dynamic and able to move from its docked position. Here, we compare DNA binding of RecBCD and a variant, RecBΔNucCD, in which the 30 kDa nuclease domain has been deleted. RecBCD binding to a blunt DNA end is enthalpically unfavorable and entropically driven. Deletion of RecBNuc results in an increase in DNA binding affinity, suggesting an allosteric effect of RecBNuc. RecBΔNucCD binding to DNA possessing fully ‘pre-melted’ DNA ends is associated with a large favorable ΔHobs, but much smaller than observed for RecBCD, suggesting that deletion of RecBNuc limits bp melting from a blunt DNA. We also solved cryo-EM structures showing only 4 bp melted upon RecBΔNucCD binding to a blunt ended DNA duplex, less than the 11 bp melted upon RecBCD binding. Thus, the RecB nuclease domain regulates the extent of bp melting by RecBCD. These results suggest that RecBNuc may manifest its long-range allosteric effect on DNA binding and DNA melting via linker-linker interactions between RecB and RecC.
|
|
Scooped by
?
December 6, 2:56 PM
|
Circular RNAs (circRNAs) are natural outputs of transcription and RNA processing in eukaryotes. Four subclasses of circRNAs have been identified in animal cells, and most circRNAs are generated via backsplicing. The intricate formation of circRNAs is orchestrated by various cis-regulatory elements and trans-acting factors. Previous studies have gained insights into the general factors and elements involved in backsplicing. Recently, modulation of circRNA biogenesis to generate tissue-specific expression patterns is coming into focus. We summarize various mechanisms involved in the biogenesis of distinct circRNA subclasses across multiple cell types. We also discuss the involvement of relevant mechanisms in human diseases and potential biomedical interventions that target circRNA pathways.
|


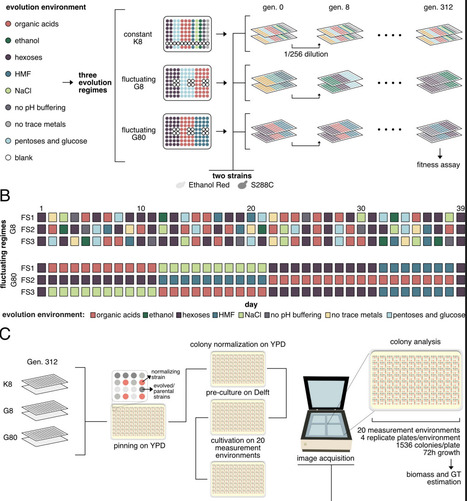

























ale, Haploids with less fit parents significantly increased in fitness, while the more-fit parental diploid strain Ethanol Red instead enhanced its robustness over time. In contrast with previous findings in yeast, evolution in fluctuating conditions improved fitness more than constant conditions in certain measurement environments.
The robustness was calculated across 20 measurement environments, using fitness data (biomass) of evolved and parental strains separately.