 Your new post is loading...

|
Scooped by
?
Today, 3:52 PM
|
Protein glycosylation is a very common post-translational modification PTM seen in all branches of biology. The functional roles for protein glycosylation are many and varied, essential in eukaryotes but seemingly dispensable in bacteria. One group of bacteria where protein glycosylation has been looked at for at least 50 years are the actinobacteria, a large and diverse group of bacteria which include well know pathogens like Mycobacteria tuberculosis, Corynebacterium diphtheriae, and well know species important in biotechnology like Streptomyces lividans and Corynebacterium glutamicum. Actinobacterial protein glycosylation is a form of protein O-mannosylation which is found widely in eukaryotes from yeast on up the evolutionary ladder but is much less understood at the functional level. Very few direct roles for protein O-mannosylation have been described in the literature. This review examines newer findings from the actinobacterial world which with the help of glycoprotein models suggests how the glycans might play a role in actinobacterial biology.
|
|
Scooped by
?
Today, 3:26 PM
|
Ammonia-oxidizing archaea (AOA) are among the most abundant microorganisms in the ocean and play a critical role in marine nitrogen cycling. Recently, urea has been shown to serve as an additional substrate for marine AOA, with substantial urea use in the ammonium-depleted open-ocean. Yet, the mechanisms that control urea use and potentially maintain high AOA abundances remain unclear. Here, we investigate urea and ammonia use by AOA in three contrasting marine environments, from coastal, ammonium-rich to open-ocean, ammonium-poor waters. Our combined results indicate that distinct substrate utilization strategies of Nitrosopumilus and Nitrosopelagicus control their environmental distribution. The more coastal AOA genus, Nitrosopumilus, primarily uses ammonium. In contrast, enhanced urea utilization in ammonium-limited waters is linked to the activity and growth of Nitrosopelagicus. Thus, the use of urea, and potentially other organic-N compounds by Nitrosopelagicus plays a major role in fueling open-ocean nitrification and sustaining primary productivity in these vast regions. Two groups of ammonia-oxidizing archaea drive marine nitrification. Stuehrenberg et al. reveal that their distribution reflects substrate use, with one relying on urea and the other on ammonia to maintain nitrification in open-ocean waters.
|
|
Scooped by
?
Today, 3:00 PM
|
Plant protein production systems are scalable and sustainable platforms capable of meeting the growing demand for functional proteins in nutrition, pharmaceuticals, and industry. Recent advances in essential amino acid (EAA) biosynthesis, gene regulation, and subcellular targeting have enhanced protein yields and stability, but are yet to be integrated into holistic engineering approaches. Metabolic engineering can improve amino acid (AA) metabolism and energy efficiency, while genetic engineering enables finetuned, spatiotemporal expression of target proteins. Coupled with in silico tools for protein design, novel proteins with enhanced stability and functionality can be developed. Integrating these strategies would enable the fine-tuning of protein synthesis while balancing cellular energy costs, offering context-dependent opportunities to advance protein production in plant systems.
|
|
Scooped by
?
Today, 2:51 PM
|
Reactive oxygen species (ROS) are a promising alternative bactericide. However, it is questioned that bacteria can potentially develop resistance to ROS, similar to their resistance against antibiotics and silver. Herein, it is reported that Gram-negative bacteria, including Pseudomonas aeruginosa, Escherichia coli, and Klebsiella pneumoniae, develop resistance to ROS after six repeated exposures. Notably, ROS minimum inhibitory concentration of P. aeruginosa significantly increases to 256-fold after ten passages. The resistance mechanism predominantly originates from the intensified biosynthesis of the highly reductive hydrogen sulfide (H2S) and pyoverdine (PVD) siderophores, effectively neutralizing ROS. Simultaneously, PVD transports Fe3+ from the extracellular space into the bacteria, releasing H2S bound to Fe3+ and enhancing ROS scavenging. Additionally, the enhanced outer membrane (OM) biogenesis establishes a robust OM barrier, impeding ROS penetration. The acquired resistance to ROS can be significantly reduced by incorporating additional Fe3+ into the culture medium or disrupting the H2S biosynthetic gene. These observations suggest that careful consideration is required when utilizing ROS against Gram-negative bacteria. It is anticipated that understanding this resistance mechanism can inform the development of future antimicrobial agents, particularly for Gram-negative bacteria.
|
|
Scooped by
?
Today, 2:31 PM
|
JBrowse 2 is an open-source genome browser that provides unique features for visualizing syntenic relationships between multiple genomes. This article describes a protocol for setting up synteny views in JBrowse 2, using an assembly-to-assembly whole-genome alignment example. We detail data preparation steps, including the generation and formatting of whole-genome alignment data into formats compatible with JBrowse 2's synteny visualization capabilities, and show the GUI-driven process for setting up interactive synteny views and generating publication-quality figures. This protocol establishes methods for using JBrowse 2 to explore conserved sequences across multiple genomes.
|
|
Scooped by
?
Today, 2:09 PM
|
Despite the importance of the cervicovaginal microbiome, the mechanisms that govern its composition and drive its impact on host physiology remain poorly understood. With the aim to expand our understanding of the function and ecology of the vaginal microbiome, we present VIRGO2, an enhanced non-redundant gene catalog comprising over 1.7 million well-annotated genes from body-site specific microbes and viruses. Analyses using VIRGO2 reveal insights such as including the identification of previously uncharacterized vaginal bacteria, features of the vaginal mycobiome and phageome, and differential expression of bacterial carbohydrate catabolic genes. Constructed from over 2500 metagenomes and 4000 bacterial genomes, VIRGO2 broadens geographic representation and microbial diversity compared to its predecessor. This updated catalog enables more precise profiling of taxonomic and functional composition from metagenomic and metatranscriptomic datasets. VIRGO2 is a critical resource for integrative analyses of vaginal microbial communities and their interactions with host tissues, thereby enhancing our mechanistic understanding of vaginal health and disease. The vaginal microbiome is a critical determinant of health. Here, the authors present VIRGO2, a gene catalog describing these microbial communities and analyze vaginal metagenomes and metatranscriptomes to derive insights into their function and ecology.
|
|
Scooped by
?
Today, 12:50 PM
|
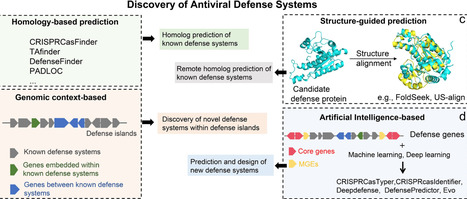
Prokaryotes possess a remarkably diverse and dynamic repertoire of antiviral defense systems, enabling them to withstand phage predation. However, their frequent horizontal gene transfer, extensive sequence diversity, modular genomic organization, and rapid evolution make purely experimental discovery challenging. Coupled with the massive influx of microbial genomes from high-throughput sequencing, computational strategies have become indispensable complementary tools that can enhance the efficiency and scope of defense systems discovery. In this review, we categorize computational approaches into four major strategies: (i) Sequence homology-based methods, which reliably annotate known defense systems through protein sequence similarity but are limited in detecting highly divergent or novel systems; (ii) Structure-guided approaches, which leverage conserved protein folds to uncover remote homologs and single-gene defense proteins, providing sensitivity beyond sequence-based identification, albeit at high computational cost; (iii) Genomic context-based strategies, which exploit gene co-localization and defense islands to uncover multi-gene defense clusters and previously uncharacterized defense modules; and (iv) Artificial intelligence-powered methods, which integrate sequence-derived embeddings with genomic context information to predict low-homology proteins and reconstruct candidate defense systems at scale, enabling discovery of novel systems beyond the reach of conventional approaches. We further discuss emerging tools and frameworks, such as the conserved gene cluster discovery tool and genomic foundation models, which hold strong potential to extend conventional approaches for identifying novel defense systems and supporting the generative design of synthetic modules. By comparing methodological principles, strengths, and limitations, this review provides a practical framework for the systematic exploration of microbial immune systems, guiding applications such as rational phage therapy, microbiome engineering, and synthetic biology.
|
|
Scooped by
?
Today, 10:49 AM
|
Plant synthetic biology holds great promise for engineering plants to meet future demands. Genetic circuits are being designed, built and tested in plants to demonstrate the proof of concept. However, developing these components in monocots, which the world relies on for grain, lags behind dicot models, such as Arabidopsis thaliana and Nicotiana benthamiana. Here, we show the successful adaptation of a ligand-inducible sensor to activate an endogenous anthocyanin pathway in the C4 monocot model Setaria viridis. We identify two transcription factors that can be expressed as a single transcript that are sufficient to induce endogenous anthocyanin production in S. viridis protoplasts and whole plants in a constitutive or ligand-inducible manner. We also test multiple ligands to overcome physical barriers to ligand uptake, identifying triamcinolone acetonide (TA) as a highly potent inducer of this system. Using hyperspectral imaging and a discriminative target characterization method in a near-remote configuration, we can non-destructively detect anthocyanin production in leaves in response to ligands. This work demonstrates the use of inducible expression systems in monocots to manipulate endogenous pigmentation production for remote detection. Applying inducible anthocyanin production coupled with sensitive detection algorithms could enable crop plants to report on the status of field contamination or detect undesirable chemicals impacting agriculture, ushering in an era of agriculture-based sensor systems.
|
|
Scooped by
?
Today, 10:25 AM
|
Chemosynthetic symbioses between animals and bacteria are known to underpin productivity in the deep sea, yet the diversity of energy and carbon sources sustaining these associations in shallow-water environments remains poorly understood. Dimethylsulfoniopropionate (DMSP) is highly abundant in coastal habitats, where it is produced by seagrasses, phytoplankton, and heterotrophic bacteria, and occurs together with its breakdown product dimethyl sulfide (DMS) in shallow-water sediments. Here we show, supported by genomic and transcriptomic evidence, that DMSP and DMS cycling are integral to the energy and carbon metabolism of the gutless oligochaete Olavius algarvensis and its chemosynthetic symbionts. By assigning DMSP degradation pathways to individual members of the host′s microbial community, we reconstructed a network integrating demethylation and cleavage with energy conservation, methionine biosynthesis, and acetate assimilation into polyhydroxyalkanoates. We also identified a host-encoded methanethiol oxidase (MtoX) suggesting host participation in MeSH detoxification. Comparative metagenomic analyses of more than 60 gutless oligochaete species from globally distributed habitats showed that key DMSP- and DMS-processing genes (dddP, dmdA, tmm, dmsA) are widespread, indicating that organosulfur metabolism is a conserved feature of these symbioses. Our findings expand the recognized metabolic repertoire of shallow-water chemosynthetic symbioses and provide evidence that these associations directly contribute to marine DMSP and DMS cycling.
|
|
Scooped by
?
Today, 10:13 AM
|
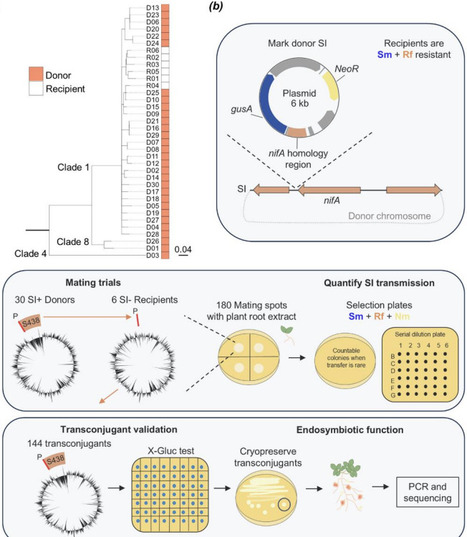
The advent of endosymbiosis underlies evolutionary innovation and ecosystem function. However, whether free-living partners tend to benefit or exploit each other during incipient endosymbiosis remains a dilemma. Rhizobia bacteria are plant endosymbionts capable of initiating root nodules and fixing nitrogen due to genes carried on mobile genetic elements (MGEs) such as the symbiosis island (SI). We conjugated marked SIs into the genomes of non-nodulating strains, which was sufficient to generate de novo root nodule-forming endosymbionts. Most novel endosymbionts originated as commensals that incurred no detectable costs to host plants, in contrast to predictions of exploitation. In fact, a third of endosymbionts originated as nitrogen fixing mutualists. Consistent with phylogenetic limits to transfer of MGE function, novel endosymbionts derived from more closely related SI donor and recipient strains showed greater nitrogen fixation. However, we did not detect phylogenetic limits to SI transmission, which could reflect selfish selection for generalized horizontal transfer of this MGE. In fact, the SI was able to displace other genomic elements residing at its characteristic tRNA gene insertion site. We thus provide genetic, genomic, and functional evidence of how MGEs can potentiate and constrain major evolutionary transitions to expand bacterial niches, with cascading effects on host organisms.
|
|
Scooped by
?
Today, 9:59 AM
|
Beneath Earth's glaciers and ice sheets lies an aquatic realm where ice, water, rock, and microbial life interact, driving chemical reactions that can collectively influence the global carbon cycle, polar oceans, and climate. Efforts to describe subglacial microbiomes have been limited by the challenge of cleanly drilling through hundreds of meters of ice, such that only a few sites have ever been directly sampled. Here we use ancient metagenomics to present the first spatiotemporal characterization of subglacial bacteria and archaea. We extracted DNA from 25 subglacial precipitate samples, sedimentary accumulations of minerals that form in subglacial waters prior to exposure on the surface. The precipitates studied here formed between 16,000 and 570,000 years ago beneath the Antarctic and Laurentide Ice Sheets. We show that postmortem DNA damage patterns can reliably distinguish between ancient subglacial and modern surface taxa, and that this approach can enable reconstruction of subglacial microbiomes across poles and ice ages. Our analysis suggests that subglacial microbiomes are dominated by chemolithoautotrophs, ultra-small microbes, and taxa closely related to those found in deep subsurface or extreme cold and hypersaline environments. These microbiomes split into two distinct clusters distinguished by oxygen availability and redox conditions, irrespective of geography or age. Geochemical measurements of subglacial redox state, measured either indirectly via precipitate calcite Fe and Mn concentrations or directly via water reduction potential, reproduce these same two clusters exactly. Our findings describe how subglacial water redox states are held in balance by microbes, hydrology, and oxygen input from fresh subglacial meltwater, that we interpret to be controlled by the ice sheet response to past climate variations.
|
|
Scooped by
?
Today, 9:41 AM
|
Diatoms are promising microorganisms to provide sustainable routes for photosynthetic terpenoid production from CO₂, yet their potential for compartmentalized engineering remains largely unexplored. Here, we systematically profiled the biosynthetic capacity of Phaeodactylum tricornutum by targeting representative synthases for hemi, mono, sesqui, and tetraterpenoids to the cytosol, chloroplast, and periplastidial compartment (PPC). This comprehensive analysis revealed that all major prenyl phosphate precursors, DMAPP, GPP, FPP, and GGPP, are accessible in all compartments, including in the PPC and can sustain heterologous flux without major physiological penalties, although production efficiency varies across compartments and product classes. By determining precursor availability, we propose the diatom PPC as a minimal engineerable organelle directly interfaced with a eukaryotic chloroplast. Moreover, we highlight its utility as a unique interface to investigate metabolic exchange between the MVA and MEP pathways. These findings provide a systematic framework for compartment-specific terpenoid engineering in diatoms and open new opportunities for modular pathway assembly and synthetic biology in photosynthetic eukaryotes.
|
|
Scooped by
?
December 5, 4:49 PM
|
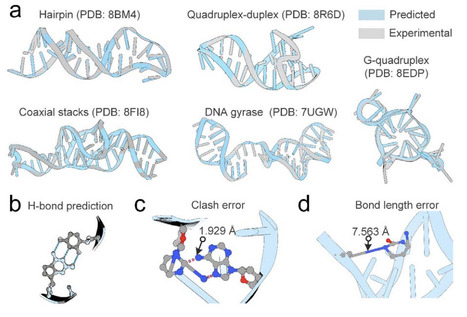
Designing biological sequences that fold into predefined conformations is a central challenge in bioengineering. Although deep learning has enabled significant advances in protein and RNA sequence design, progress in single-stranded DNA (ssDNA) design has been constrained by the limited availability of structural data. To address this challenge, we introduce InvDNA, a deep learning-based method that designs ssDNA sequences directly from backbone atomic coordinates. This end-to-end formulation avoids the loss of structural information during backbone-to-feature conversion and further accommodates flexible backbone representations, dynamic sequence masking, and structural reconstruction objectives. All these training strategies enhance the capacity of InvDNA to generalize across diverse ssDNA structural contexts while enabling additional functionalities, including generating diversity sequences for a given backbone, reconstructing base conformations from backbone and preserving functional sites. In benchmarks using experimentally determined ssDNA structures, InvDNA achieves over a twofold improvement in sequence recovery compared with existing ssDNA design approaches. Further computational validation using AlphaFold3 shows that 44.4% of InvDNA-designed sequences successfully fold into their predefined conformations. Notably, this success rate increases when backbone coordinates are perturbed to diversify the InvDNA-designed sequences. Collectively, these results establish InvDNA as a robust framework for rational ssDNA engineering.
|
|
|
Scooped by
?
Today, 3:39 PM
|
Type VI secretion systems (T6SSs) are molecular machines used by bacteria to release effectors that target either host cells, competing bacteria or fungi. Regulatory mechanisms underlying antifungal T6SS activity remain unexplored. Here we show, using mouse infection with wild-type and T6SS mutant bacteria, that T6SS activity of the enteropathogen, Yersinia pseudotuberculosis (Yptb), reduces fungal prevalence in the gut microbiota and has direct activity on Candida albicans. Screening of bacterial effector mutant strains, and structural and biochemical analyses identify TfeC as an antifungal chitinase T6SS effector that can kill C. albicans. In vivo experiments confirm that TfeC expression promotes Yptb colonization and reduces C. albicans abundance. We also show that Yptb senses the fungal quorum-sensing molecule, tyrosol, through the two-component system, EnvZ–OmpR, and responds by activating T6SS4. Our findings suggest that Yptb modulates its antifungal activities by detecting changes in fungal population density cues, revealing a mechanism of fungal–bacterial interkingdom communication mediated by fungal quorum-sensing molecules. Yersinia pseudotuberculosis senses fungal tyrosol signalling through EnvZ–OmpR which triggers T6SS activation and antifungal effector release to reduce fungal competitors in the mouse gut.
|
|
Scooped by
?
Today, 3:16 PM
|
E. coli RecBCD, a hetero-trimeric helicase and nuclease, functions in double stranded (ds) DNA break repair. RecBCD possesses ATPase motor domains within both RecB (3’ to 5’) and RecD (5’ to 3’) and a nuclease domain within RecB (RecBNuc). RecBCD binds to double stranded DNA ends and initiates DNA unwinding by first melting several DNA base pairs (bp) using only its binding free energy. The RecBNuc domain is docked ∼70 Å from the duplex DNA binding site in RecBCD-DNA structures but appears to be dynamic and able to move from its docked position. Here, we compare DNA binding of RecBCD and a variant, RecBΔNucCD, in which the 30 kDa nuclease domain has been deleted. RecBCD binding to a blunt DNA end is enthalpically unfavorable and entropically driven. Deletion of RecBNuc results in an increase in DNA binding affinity, suggesting an allosteric effect of RecBNuc. RecBΔNucCD binding to DNA possessing fully ‘pre-melted’ DNA ends is associated with a large favorable ΔHobs, but much smaller than observed for RecBCD, suggesting that deletion of RecBNuc limits bp melting from a blunt DNA. We also solved cryo-EM structures showing only 4 bp melted upon RecBΔNucCD binding to a blunt ended DNA duplex, less than the 11 bp melted upon RecBCD binding. Thus, the RecB nuclease domain regulates the extent of bp melting by RecBCD. These results suggest that RecBNuc may manifest its long-range allosteric effect on DNA binding and DNA melting via linker-linker interactions between RecB and RecC.
|
|
Scooped by
?
Today, 2:56 PM
|
Circular RNAs (circRNAs) are natural outputs of transcription and RNA processing in eukaryotes. Four subclasses of circRNAs have been identified in animal cells, and most circRNAs are generated via backsplicing. The intricate formation of circRNAs is orchestrated by various cis-regulatory elements and trans-acting factors. Previous studies have gained insights into the general factors and elements involved in backsplicing. Recently, modulation of circRNA biogenesis to generate tissue-specific expression patterns is coming into focus. We summarize various mechanisms involved in the biogenesis of distinct circRNA subclasses across multiple cell types. We also discuss the involvement of relevant mechanisms in human diseases and potential biomedical interventions that target circRNA pathways.
|
|
Scooped by
?
Today, 2:44 PM
|
Amyloids, once viewed solely as pathological hallmarks, are now recognized as widespread and versatile functional protein assemblies. Bacterial functional amyloids (FuBAs), particularly curli (CsgA) from Escherichia coli and FapC from Pseudomonas, have emerged as paradigms for understanding amyloid structure, assembly, and function. The recent cryo-EM-based structure of FapC, together with others’ combined cryo-EM and integrative computational studies on CsgA, reveal a β-solenoid fold stabilized by imperfect repeats, producing fibrils of exceptional stability and low polymorphism, whose biogenesis is tightly controlled through dedicated accessory factors, ensuring precise secretion and nucleation. FuBAs not only scaffold biofilms but also display intrinsic catalytic activity, expanding the biochemical repertoire of extracellular matrices. They also exhibit hierarchical mechanical properties ranging from GPa stiffness at the fibril core to kPa elasticity in hydrated biofilms. FuBA operons are phylogenetically widespread, with repeat variation contributing to sequence diversity and functional adaptability. FuBAs might be seen as evolutionary intermediates between disordered peptides with significant self-interaction tendencies and highly structured globular proteins. Their simple structures make them robust platforms for biomaterial engineering. Understanding the interplay between sequence repeats, fibril architecture, and emergent functions opens avenues for harnessing amyloids as programmable nanomaterials with applications in catalysis, synthetic biology, and biofilm control.
|
|
Scooped by
?
Today, 2:19 PM
|
Tools to edit DNA methylation in a targeted manner are vital for establishing causal relationships between DNA methylation and its function, as well as for plant breeding and gene therapy. Here, by constructing dCas9 fusions to a panel of effectors and cofactors, we develop a range of highly effective tools for editing DNA methylation in Arabidopsis, including five tools for DNA methylation and six tools for DNA demethylation. Our tools show a diversity of performance features in terms of specificity and efficiency, offering either the capacity to edit DNA methylation in a target-specific manner or the ability to edit DNA methylation genome-wide due to potent off-target effect. Importantly, DNA methylation edited by these tools is inherited in the absence of transgene. These versatile tools pave the way for diverse applications of DNA methylation editing in not only research but also epigenetic breeding of crops. Targeted DNA methylation editing is critical for establishing the causal relationship between DNA methylation and its function as well as for epigenetic crop breeding. Here, the authors develop a CRISPR/dCas9-based fusion protein strategy for DNA methylation and demethylation editing in Arabidopsis.
|
|
Scooped by
?
Today, 1:10 PM
|
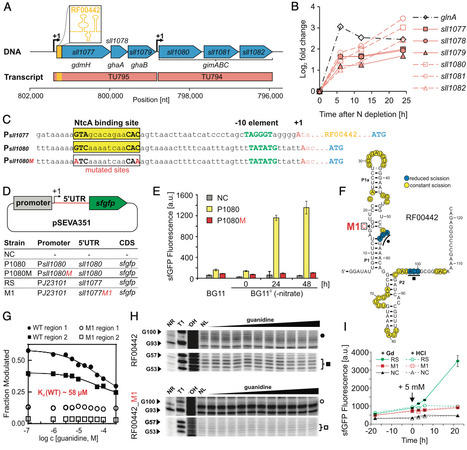
Guanidine is well known as a denaturing agent. However, recent studies have demonstrated both the widespread synthesis of guanidine, e.g., in plants and mammals, as well as the widespread occurrence of guanidine metabolism in bacteria, suggesting a broader biological role. Here, we provide insights into guanidine assimilation via guanidine hydrolases (GdmH) in cyanobacteria. The gdmH gene is widespread among cyanobacteria and enables growth on guanidine as the sole nitrogen source. Consistent with this, gdmH gene expression increased under nitrogen limitation, regulated by the transcription factor NtcA. However, guanidine is toxic above 5 mM, necessitating GdmH activity and adaptive mutations activating the multidrug efflux system PrqA. The gdmH gene is frequently colocalized with ABC transporter genes (named gimABC), which are driven by an additional NtcA-regulated promoter. The corresponding substrate-binding protein GimA showed high affinity to guanidine. Consistent with a high affinity import system, disruption of genes gimA or gimB impaired guanidine-dependent growth of Synechocystis sp. PCC 6803 at low concentrations. However, in presence of >1 mM guanidine, these mutants grew like wildtype, suggesting the existence of additional uptake mechanisms for guanidine. We also demonstrate the high-affinity binding of guanidine to a previously described, conserved RNA motif located within the gdmH 5’-untranslated region, validating it as a guanidine-I riboswitch. By combining it with various promoters, we achieved precise, titratable control of heterologous gene expression in cyanobacteria in vivo. Our findings establish guanidine assimilation as an integral element of cyanobacterial nitrogen metabolism and highlight guanidine riboswitches as valuable tools for synthetic biology.
|
|
Scooped by
?
Today, 10:56 AM
|
The escalating crisis of antimicrobial resistance demands novel therapeutics that overcome the limitations of conventional antibiotics. Human α-defensins, such as Human Neutrophil Peptide-1 (HNP-1), represent compelling candidates due to their potent, broad-spectrum antimicrobial activity and membrane-disrupting mechanism. However, clinical translation has been hindered by the absence of scalable production systems capable of delivering high yields of functional peptide. To address this challenge, we developed an efficient expression platform in the yeast Komagataella phaffii. In this system, a codon-optimized HNP-1 sequence was fused to a His6-SUMO tag downstream of the α-factor secretion signal to enhance solubility, folding, and recovery. Initial shake-flask optimization identified pH 6.0 with 96 h of induction as optimal fermentation conditions, yielding 19.75 ± 1.1 mg/L of recombinant protein. Subsequent scale-up to a controlled 5 L bioreactor significantly enhanced production, achieving 122 mg/L of the fusion protein. Following purification and precise cleavage, this process delivered 15.25 mg/L of pure, mature HNP-1 representing, to our knowledge, the highest yield of recombinant HNP-1 reported. The final product demonstrated potent antibacterial activity against Staphylococcus aureus and Escherichia coli while exhibiting excellent hemocompatibility, confirming preservation of native structure and function. This work establishes a scalable production platform for HNP-1 and provides an adaptable framework for expressing other structurally complex antimicrobial peptides with therapeutic potential.
|
|
Scooped by
?
Today, 10:33 AM
|
Techniques for selecting or sorting single cells within large populations of genetic variants are central to synthetic biology and biotechnology. Widely-used methods such as Fluorescence Activated Cell Sorting (FACS) enable rapid processing of large libraries, but are restricted to low-dimensional measurements taken at a single time point. As a result, sorting based on dynamic or multi-trait phenotypes---such as transient properties and properties that occur on fast timescales or in response to dynamic actuating signals---remains fundamentally challenging. Here we introduce Microscopic PhotoSelection (MiPS) which employs an automated robotic platform for single-cell selection based on multiple dynamic criteria, directly on microfluidic mother machine devices. The system couples long-term single-cell imaging with real-time analysis and selective optical targeting, allowing fully automated enrichment using high-intensity UV light or alternative wavelengths with the addition of photosensitisers. By targeting many cells in parallel, our platform overcomes throughput limitations of existing microfluidic-based selection technologies such as optical tweezers or droplet-based methods and provides a direct approach to select cells based on time-resolved, multi-trait phenotypes. We demonstrate the ability to perform in vivo selection and outline how iterative, feedback-based selection strategies can refine enrichment across multiple rounds. Taken together, our work establishes a high-throughput selection framework integrated into microfluidic devices, enabling applications in directed evolution, biosensor optimisation, circuit engineering, and diagnostics, where selection based on dynamic, multi-trait phenotypes is essential.
|
|
Scooped by
?
Today, 10:21 AM
|
Detection of morphological phenotypes from light microscopy is a key part of microbiology. Despite advances in automated morphological analysis, accurate measurements still often require significant user input. To address this, we have developed micromorph, a Python package to measure bacterial morphological properties from multiple types of light microscopy data. micromorph is available as a Python package with a documented API, or as napari plugin, thus appealing to a range of users regardless of their coding proficiency. The micromorph API allows easy integration into custom analysis pipelines. We demonstrate a range of scenarios in which micromorph outperforms currently available bacterial morphology algorithms and introduce a new approach for reliably measuring the widths and lengths of quasi-circular cells.
|
|
Scooped by
?
Today, 10:03 AM
|
Many clinically important antimicrobial resistance (AMR) phenotypes such as fluoroquinolones and rifamycins are driven by antimicrobial resistance-conferring mutations (ARMs) in conserved chromosomal loci (e.g., gyrA, parC, rpoB). Resistome profiling by metagenomics sequencing is often proposed as an ideal AMR surveillance tool as it is organism-agnostic, but currently, existing metagenomic AMR surveillance pipelines are only able to identify acquired AMR genes but not point mutations associated with AMR. This is a serious gap in metagenomics-based AMR surveillance, as the true extent of AMR may be underestimated. We developed MetaPointFinder (v1), a read-based method that can process both long and short metagenomic reads. The tool identifies resistance-determining regions in reads using DIAMOND (translated protein) and KMA (nucleotide), and classifies known resistant versus wild-type variants by aligning the sequences using pwalign and assessing the observed mutations based on known resistance mutations in the AMRFinderPlus database. The tool outputs ARMs per read, per gene and per antibiotic class. In proof-of-concept analyses, MetaPointFinder identified known AMR-associated mutations and quantified resistant/susceptible read counts and ratios from metagenomic samples with corroborating phenotypic resistance data when available. We benchmark our tool using simulated reads from DNA and from reverse-translated protein references with read lengths of 100-5000 bp and error rates between 0 and 30%, simulating both Illumina and Nanopore error rates. We show that MetaPointFinder outperforms any available method for detection of ARMs in metagenomics data. MetaPointFinder complements gene-centric resistome profiling by capturing chromosomal mutation-based AMR directly from metagenomes.
|
|
Scooped by
?
Today, 9:50 AM
|
Genomes encode the instructions for life, yet their full interpretation requires models capable of capturing long-range context and functional meaning at scale. Existing genome language models (gLMs) are limited by short context windows, high computational cost, and poor interpretability. We present GenSyntax, a product-contextualized large language model (LLM) trained on 49,250 annotated prokaryotic genomes. GenSyntax replaces nucleotide tokenization with gene product descriptors, transforming genomes into "genetic paragraphs" that preserve functional semantics. Using a two-stage training strategy, GenSyntax achieves leading performance in plasmid host identification, gene function prediction, genome assembly, and gene essentiality assessment compared with the other LLMs. It also enables phenotype prediction and minimal genome design, establishing a scalable and interpretable framework for genome-scale decoding and synthetic biology.
|
|
Scooped by
?
December 5, 4:52 PM
|
Starships are a recently discovered superfamily of extremely large mobile genetic element (MGE)s in fungi that encode diverse gene sequences, many of unknown function. Starships are widespread throughout filamentous Ascomycetes (Pezizomycotina), but relatively little is known about their fine-grained distributions at lower taxonomic levels. As more and more Starships are discovered, it is increasingly important to more effectively catalog their gene contents and taxonomic distributions to better understand their contributions to fungal evolution. To address this, we developed starbase, a web server and comparative toolkit for exploring Starship diversity and hypothesis generation. The starbase database is constructed from Starships identified from existing studies, as well as an exhaustive de novo survey of Starship sequences within a set of 19 863 publicly available fungal genome assemblies. This database consists of 5 493 Starships, their associated nucleotide sequences, captain gene protein sequences, cargo gene annotations, and other metadata pertaining to the annotation and analysis of Starships in fungal genomes. As a resource, starbase provides new avenues for studying structural variation in fungal genomes. starbase provides several key features for the research community: a centralized repository of curated Starship annotations, a standardized accessioning system enabling consistent referencing of elements across studies, tools for searching existing sequences and classifying novel Starships based on established classification schemes, and a submission portal encouraging community contributions. As Starship identification becomes a routine component of fungal genome annotation, starbase provides a framework for organizing this growing body of data and facilitating comparative analyses across the expanding landscape of fungal genomic diversity.
|




























guanidine sensor