 Your new post is loading...

|
Scooped by
mhryu@live.com
Today, 1:47 AM
|
Bacteria are constantly exposed to changing environmental conditions, and to survive they need to adapt quickly, adjusting their gene expression and metabolism to make the most of the resources available. One of the mechanisms involved is the stringent response, characterized by production of specific guanosine derivatives—ppGpp and pppGpp (collectively called (p)ppGpp). These regulators exert their action and coordinate a global response at many different levels, for example, transcription, translation, nucleotide metabolism, DNA replication, and carbon and lipid metabolism. In this review, we discuss how (p)ppGpp is synthesized and degraded, how it controls different cellular processes and their interplay with other second messengers. A description of differences in (p)ppGpp regulation in Gram-negative and Gram-positive bacteria, along with recent findings and some historical perspectives, is provided. We argue that although much is known about the stringent response and the novel discoveries are definitely advancing the (p)ppGpp field, they are not exhaustive. Instead, they seem to constantly point to aspects that are still waiting to be uncovered—thus, the (p)ppGpp magic goes on.
|
|
Scooped by
mhryu@live.com
Today, 1:41 AM
|
Phenazines are small, redox-active secondary metabolites produced by various bacterial species. These compounds participate in electron-transfer reactions, aiding microbes in surviving stressful or oxygen-limited environments. In this review, we examine the extensive structural diversity of phenazines and trace the evolutionary history of their biosynthetic pathways, which often move between distantly related species through horizontal gene transfer. We also explore how environmental factors such as nutrient levels and cell-to-cell signaling regulate phenazine production. Beyond their roles in microbial physiology, phenazines influence interactions among organisms, acting as antimicrobial agents, signaling molecules, and factors that shape microbiome dynamics in soils, plant roots, and other habitats. A better understanding of phenazine biology reveals how microbes adapt and thrive in diverse environments and emphasizes the potential applications of these compounds in agriculture and human health.
|
|
Scooped by
mhryu@live.com
Today, 1:28 AM
|
Most known chemicals originate from humans, thousands enter industrial usage annually, and new chemicals pose a continuous challenge to microbial evolution. The evolution of microbes to biodegrade new chemicals is crucial in protecting human and ecosystem health. New chemical biodegradation requires the evolution of new enzymes and metabolic pathways to meet the challenge. The rate of this process is determined by the structures of the new chemicals and preexisting enzymes, and the available metabolic pathways of the host microbe. Existing metabolism evolved over billions of years in response to naturally occurring chemicals. Natural petroleum is one example. Its diverse chemical structures have provided a training ground for microbial evolution. Similarly, studies on the biodegradation of petroleum have elucidated mechanisms that microbes have recruited to degrade industrial chemicals. Such studies have also led to the concepts of co-oxidation, co-metabolism, and enzyme promiscuity, which underlie new enzyme evolution. The focus of the present review is on evolutionary adaptations leading to the microbial biodegradation of non-polymeric industrial organic molecules. The greatest challenges to microbes and evolution are chemicals synthesized to resist biodegradation. A major current example is for per- and polyfluorinated alkyl substances, often known as PFAS. Most recently, directed evolution and artificial intelligence are being applied to the problems posed by highly resistant chemicals.
|
|
Scooped by
mhryu@live.com
Today, 1:12 AM
|
Adaptation of Escherichia coli to osmotic upshift requires rapid accumulation of intracellular solutes to restore turgor and maintain cellular homeostasis. While compatible solutes are well-established contributors to this process, they do not fully account for the early events following osmotic stress. Here, we demonstrate that inorganic phosphate and phosphorylated metabolites play a major and previously underappreciated role in osmoadaptation. Following osmotic upshift under conditions where accumulation of compatible solutes is restricted, E. coli exhibits a substantial increase in intracellular phosphate after a short lag. This increase accounts for a significant fraction of the charge balance required during rapid uptake of K⁺ and NH₄⁺, the latter supporting glutamate synthesis as a principal counterion. Concomitantly, nucleotide pools display complex, multiphasic dynamics, including a transient decrease in adenylate energy charge whose duration correlates with stress magnitude. In addition, levels of pyrophosphate and key glycolytic intermediates, including dihydroxyacetone phosphate and 1,3-bisphosphoglycerate, increase markedly, indicating redistribution of phosphate into central metabolic pathways. These findings support a model in which phosphate uptake and metabolic redistribution contribute both to intracellular charge balance and to dynamic metabolic reorganisation during osmotic stress. By linking ion transport with central metabolism, this work expands current models of bacterial osmoadaptation and identifies phosphate flux as a key component of the early stress response.
|
|
Scooped by
mhryu@live.com
Today, 12:41 AM
|
Efficient intracellular protein delivery represents an essential prerequisite for protein-based biotechnologies and therapeutics targeting intracellular components. However, this process is limited by multiple factors, including nonspecific protein binding, insufficient cellular uptake, inefficient endosomal escape, and inadequate cytosolic protein release. Here we show that by engineering fully recombinant supercharged protein nanocages, we achieve exceptionally high cellular uptake using a strategy we term ‘supercharged interface engineering’. By incorporating unnatural amino acids bearing phenylboronic acid groups, we develop a representative protein nanocage, pFn + . Simply mixing pFn+ with protein cargoes forms a noncovalent complex possessing enhanced cellular uptake efficiency, robust endosomal escape capability, and excellent biocompatibility. Notably, this system successfully delivers functional gene-editing tools and therapeutic antibodies in female mouse models. These findings indicate that pFn+ represents a promising platform for enhancing the cytosolic delivery of protein cargoes. Moreover, the proposed supercharged interface engineering strategy is valuable for advancing next-generation intracellular protein delivery systems. Protein therapeutics require efficient intracellular protein delivery. Here, the authors report on supercharged ferritin nanocages, loaded by simple mixing, which allow for cytosolic protein delivery through supercharged interface engineering, demonstrating application for gene editing and antibody delivery.
|
|
Scooped by
mhryu@live.com
Today, 12:10 AM
|
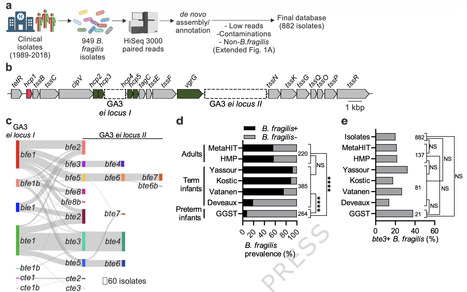
Bacteroides fragilis is a common member of the human colonic microbiota. In children, the B. fragilis Type 6 Secretion System (T6SS) has been suggested to mediate dominance between coexisting strains. We sequenced nearly 900 clinical isolates and characterized their T6SS toxin repertoire. The T6SS effector Bte3 was identified as one of the most prevalent effectors within this human isolate cohort. Bte3 exhibited the ability to intoxicate both B. fragilis and Enterobacteriaceae in vitro, and was defined as a periplasmic-intoxicating effector that employs previously unknown adaptor proteins for its secretion. The role of bte3 in early life competition was interrogated via a murine model in which neonates were exposed upon birth to two T6SS-isogenic strains. We observed that T6SS-engineered strains and human isolates containing bte3-based toxin combinations displaced competing B. fragilis strains unless these strains manifest immunity to bte3. These studies suggest that the landscape of T6SS effectors in the human population may be shaped not only by temporal exposure to distinct strains, but by potency of effector function. Our work underscores the relevance of T6SS diversity in B. fragilis early life competition and provides a potential roadmap to design microbial interventions in the context of neonatal acquisition of colonic commensals. In this study, authors show that the human gut bacterium Bacteroides fragilis uses a ubiquitous antibacterial toxin, Bte3, to eliminate diverse competing bacteria, providing a crucial competitive advantage during the initial assembly of the neonatal microbiome.
|
|
Scooped by
mhryu@live.com
June 8, 6:44 PM
|
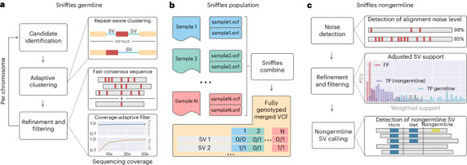
Structural variants (SVs) are the most common nucleotide alteration per human genome compared to other variant types and have profound implications in evolution, diseases and regulation of genes. Sniffles2 (v2.6.3) is an open-source software for reliable detection of SVs ranging from 50 bp and upward across deletions, duplications, insertions, inversions and translocations, based on long-read sequencing. The tool is commonly used for various species and has identified multiple causative mutations in human diseases, key alleles in cancer progression. Here this protocol describes how to fully use Sniffles2 to accurately identify germline and mosaic SVs. The latter are low-variant allele fraction SVs (5–22% VAF) of parts of cells. We further provide detailed instruction for SV joint calling within Sniffles2 to enable tumor/normal calling or family trio analysis, which can scale up into large-scale population studies. Sniffles2 has been benchmarked and compared to currently available tools and has the highest precision while also being very fast, able to call SVs for a 40× human genome in ~34 CPU min (~8.5 min wall clock with the default four threads). It is designed to be easy to use for researchers with basic knowledge of Linux operating systems and the terminal. Sniffles2 is an open-source software for reliable detection of structural variants (50 bp and upward) from long-read sequencing. Sniffles2 is able to call variants over a wide variant allele fraction (5–100%) and across multiple samples (population).
|
|
Scooped by
mhryu@live.com
June 8, 6:27 PM
|
Extracellular proteases are important signaling molecules in coagulation, inflammation, cell migration, and pain. Dysregulation of extracellular protease activity is common in diseases that perturb these critical functions. Engineering cells to sense and respond programmatically to protease activity has applications in biosensing, cell-based screening for protease activity, and therapeutics. Here we report synthetic protease-activated receptors (SynPARs) based on engineered, auto-inhibited G protein-coupled receptors (GPCRs). Relief of autoinhibition by proteolysis enables receptor activation by an exogenous or tethered agonist to generate transgene expression, real-time fluorescence, or endogenous G-protein signaling. We demonstrate SynPAR modularity with diverse secreted proteases, establish a cell-based SynPAR library selection to optimize protease recognition sequences, and control neuronal activity in response to protease activity. Finally, we use SynPARs in the dorsal root ganglion of mice to counteract hyperalgesia produced by trypsin activity, rewiring neurons to produce an analgesic response to a pain-inducing stimulus. Our study establishes SynPAR as a versatile and modular platform for recording, sensing, and responding to pericellular proteolysis. This fills a critical gap in protease-sensing tools and lays the groundwork for protease-activated genetic and cell-based medicines.
|
|
Scooped by
mhryu@live.com
June 8, 6:09 PM
|
Consortia of microbial isolates, also known as synthetic communities (SynComs), are increasingly used to study and harness microbe-microbe and microbe-host interactions. Since “synthetic” potentially evokes negative connotations, we propose adopting the term “Defined Microbial Community” for practical applications.
|
|
Scooped by
mhryu@live.com
June 8, 12:00 PM
|
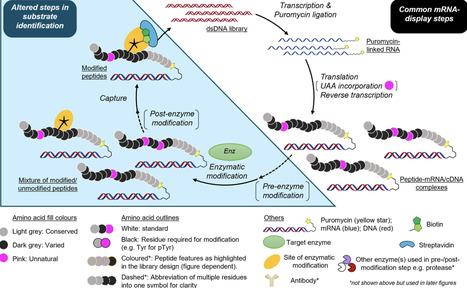
Among display technologies, mRNA display has emerged as a powerful approach to identify de novo ligands for proteins of interest. Applying the same methods to studying the substrates of protein/peptide-modifying enzymes has received much less attention, but progress in this area has accelerated rapidly over the last 5 years. In this article, we review the published literature to date (up to December 2025) of dedicated efforts to identify and understand enzyme substrate preferences using mRNA- or cDNA-display. We include observations of trends from these reports over time and reflections on where the area may go in the future. We hope this will serve as a useful primer for researchers in this and related areas.
|
|
Scooped by
mhryu@live.com
June 8, 11:55 AM
|
The large serine recombinase Bxb1 catalyzes recombination between DNA molecules containing compatible attP and attB sequences, offering broad applications in genome engineering and gene therapies. Here, we present cryo-electron microscopy structures of the Bxb1-attP-attB synaptic complex in four distinct functional states during its recombination cycle. Notably, the Bxb1 complex structures in the pre-, mid-, and post-strand-exchange states explain how the attP- and attB-bound Bxb1 dimers are assembled into a tetrameric synaptic complex and how an approximately 180° rotation occurs between the left and right dimers after DNA cleavage, thereby enabling DNA strand exchange and religation. Furthermore, we engineered Bxb1 variants with altered DNA preferences and enhanced recombination activity, which improved programmable gene integration in human cells. Overall, our findings advance the mechanistic understanding of large serine recombinases and provide a structural framework for future engineering of Bxb1-mediated genome integration technologies.
|
|
Scooped by
mhryu@live.com
June 8, 11:37 AM
|
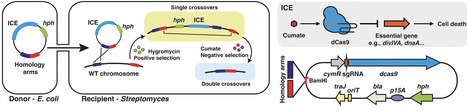
Streptomycetes are prolific producers of bioactive natural products, but many of the biosynthetic gene clusters (BGCs) are silent in the laboratory. Genetic manipulation is important to unlock their full potential. CRISPR–Cas-based genome editing has greatly advanced genetic engineering in Streptomyces. However, several challenges remain, including Cas nuclease toxicity, unintended genomic rearrangements, and elimination of the delivery plasmid. Here, we present a novel genome editing strategy that harnesses cumate-inducible CRISPR interference (CRISPRi) to transiently knockdown essential genes such as divIVA or dnaA as counterselectable marker. This enforces loss of the vector backbone, promotes homologous recombination, and yields markerless mutants by loss of the antibiotic resistance cassette during the final recombination step. We demonstrate the versatility of the ICE system (Inducible CRISPRi targeting an Essential gene) by (i) deleting four BGCs in Streptomyces coelicolor M145, (ii) inserting both a promoter and a large BGC, and (iii) introducing precise single-nucleotide substitutions. Furthermore, deletion of the prodigiosin BGC elicited expression of a poorly expressed BGC for prolinolexin lipopeptides in Streptomyces roseifaciens DSM 106196T. Considering that different essential genes may be targeted, we anticipate that inducible CRISPRi-based counterselection may be adaptable to genome editing strategies in a broad range of microbial systems.
|
|
Scooped by
mhryu@live.com
June 8, 11:28 AM
|
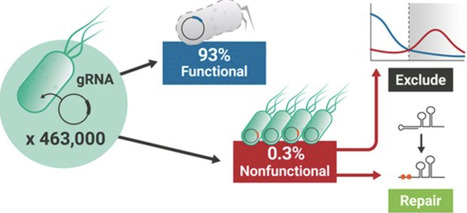
The Cas9 nuclease has become central to modern methods and technologies in synthetic biology, largely due to the ease with which it can be targeted to specific DNA loci via guide RNAs (gRNAs). Reports vary widely on the actual specificity of this targeting, with some studies observing 60% of gRNAs possessing no activity against the genome, yet an assumption persists within the E. coli community that inactive gRNAs are rare. To resolve these contradictions, we evaluated the activity of 463 000 unique gRNAs in the E. coli K12 MG1655 genome. We show that the overwhelming majority (at least 93%) of unique gRNAs are functional while only 0.3% are nonfunctional. These nonfunctional gRNAs exhibit strong spacer self-interaction, which can either be excluded using a simple design rule or “repaired” during library design. Finally, this work provides the greater microbial synthetic biology community both a set of nearly half a million empirically evaluated E. coli gRNAs as well as a thoroughly evaluated experimental procedure, complete with appropriate controls for Cas9 activity, for conducting Cas9 assays in E. coli specifically and bacteria more generally. Lastly, we have produced a webapp to allow users to easily browse and extract gRNA sequences from the E. coli genome, which can be accessed at https://grna.ornl.gov.
|
|
|
Scooped by
mhryu@live.com
Today, 1:45 AM
|
Glutamate is an essential building block and the most important amino group donor in the cell. The reactions involved in synthesis and degradation link carbon to nitrogen metabolism. The synthesis and activity of the enzymes catalyzing these reactions must, therefore, be precisely regulated. In the Gram-positive model bacterium Bacillus subtilis, glutamate is exclusively synthesized by the combined action of the glutamine synthetase (GS) and the glutamate synthase (GOGAT). The GS catalyzes the ATP-dependent assimilation of ammonium, resulting in the formation of glutamine. Glutamine is converted together with 2-oxoglutarate by the GOGAT into glutamate, which can be used either for further assimilation of ammonium, or as a building block, or amino group donor. The glutamate dehydrogenases (GDHs) GudB and RocG are strictly devoted to glutamate degradation. In recent years, exciting new observations have been made in nitrogen metabolism in B. subtilis. The GS, GOGAT, and the GDHs are multifunctional enzymes, with the GS and GDHs acting as trigger enzymes in the control of gene expression, in addition to their enzymatic activity. The glutamate-synthesizing GOGAT acts as a counter enzyme, inactivating the major GDH GudB to prevent a futile cycle. In this review, we intend to summarize the current state of knowledge about nitrogen metabolism in B. subtilis and discuss open questions that need to be answered in the future.
|
|
Scooped by
mhryu@live.com
Today, 1:39 AM
|
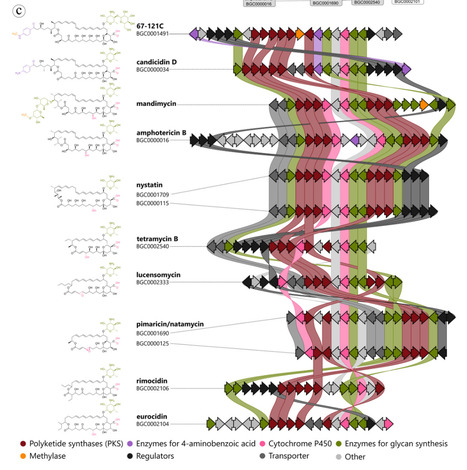
The high mortality associated with invasive fungal infections, coupled with limited therapeutic options, underscores the urgent need for antifungal agents with novel mechanisms of action. Natural products represent a particularly valuable resource, providing structurally diverse and evolutionary refined scaffolds that often outperform those found in synthetic libraries. Historically, microorganisms have proven to be a rich source of antibiotics and other therapeutic agents. Access to diverse phylogenetic lineages and biosynthetic pathways has been essential for antifungal drug development. In this review, we highlight the chemical and biosynthetic diversity of antifungal natural products derived from both fungi and bacteria. We emphasize that microbial natural products continue to play a crucial role in antifungal development, particularly through the integration of natural product chemistry, microbiology, genetics, and advanced omics technologies.
|
|
Scooped by
mhryu@live.com
Today, 1:24 AM
|
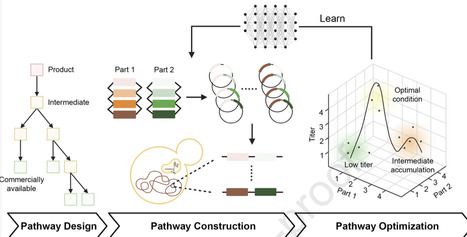
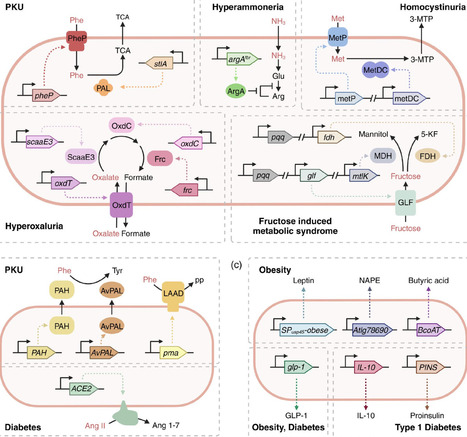
Metabolic diseases, such as obesity and diabetes, have risen due to lifestyle changes. Traditional treatments, including dietary modifications and pharmacological interventions, are limited by low compliance and adverse effects, highlighting the need for alternative therapeutic approaches that offer improved patient compliance and long-term effectiveness. Engineered live biotherapeutic products (eLBPs) have emerged as a promising strategy that combines bacterial chassis with synthetic genetic circuits for precise and targeted disease treatment. Unlike conventional therapeutics, eLBPs can colonize the intestinal tract and enable localized and condition-responsive therapeutic activity while offering improved safety profiles through defined mechanisms of action. This review highlights key strategies for eLBP development, particularly chassis selection and genetic circuit design. Applications in metabolic diseases, including inherited disorders such as phenylketonuria (PKU), demonstrate how engineered gene circuits can modulate specific metabolic pathways. However, several challenges remain, including genetic stability, interindividual variability, biological safety, and production scalability. In addition, further research on host–microbiota interactions is required to improve therapeutic predictability and efficacy, supporting the development of safe and effective personalized eLBP-based therapies for metabolic diseases.
|
|
Scooped by
mhryu@live.com
Today, 1:00 AM
|
Generative protein language models (pLMs) enable exploration of vast sequence spaces for protein design, but reliably controlling generation toward desired functional families remains challenging. While protein generation has broadly followed trends in NLP, two directions remain underexplored: alignment methods that optimize model behavior toward design objectives, and prompting-based control at inference time without fine-tuning. We introduce ProtGPT3, an open-source family of protein language models spanning 112M to 10B parameters and integrated with the Hugging Face ecosystem. The suite includes both single-sequence and multiple sequence alignment (MSA)-promptable models, enabling flexible conditioning for generation. Across model scales and control settings, we systematically compare supervised fine-tuning and few-shot prompting using homologous sequences. Analogous to how large language models (LLMs) are routinely aligned with user intent, we study post-training alignment in single-sequence models using sequence-complexity and structure-confidence metrics across the proteome. We find that alignment reduces low-complexity generations while preserving sequence diversity. Furthermore, we show that few-shot prompting is a competitive and more scalable alternative to supervised fine-tuning for controlled generation. In a low-data defluorinase case study, ProtGPT3-MSA achieved higher computational success rates than fine-tuned baselines and produced designs that were soluble and expressed following experimental validation. Finally, we explore the potential of inference-time compute in MSA models by introducing a homolog-based Feynman--Kac inference procedure for steering protein generation toward desired targets. We make our models publicly available at https://huggingface.co/collections/AI4PD/protgpt3-family .
|
|
Scooped by
mhryu@live.com
Today, 12:20 AM
|
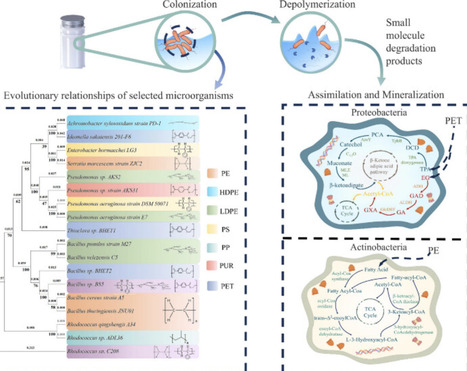
Plastic pollution is a major environmental challenge, given its widespread presence and potential risks to both ecosystems and human health. Microbial biodegradation offers a promising solution for managing plastic waste. However, research in this area remains fragmented, with varying reports on degradation efficiency, mechanisms, and practical applications. This review synthesizes recent developments in microbial taxonomy, enzymatic depolymerization, and multiomics functional analysis, highlighting the connections between microbial traits and the four key stages of plastic degradation: colonization, depolymerization, assimilation, and mineralization. It critically evaluates the robustness and comparability of reported degradation metrics, demonstrating that variability in polymer properties, experimental conditions and assessment methods severely hinders meaningful comparisons across studies and limits the translation of laboratory findings to real-world applications. Additionally, this review elaborates on emerging bioaugmentation strategies such as genetic modification, enzyme engineering and synthetic microbial consortia design, while identifying key translational bottlenecks involving scalability, environmental relevance and long-term stability. By integrating insights from multiomics research and synthetic biology, this work proposes a framework to bridge the gap between laboratory-based discoveries and practical biodegradation strategies. It aims to advance microbial plastic remediation research by identifying key knowledge gaps and offering actionable recommendations for future developments.
|
|
Scooped by
mhryu@live.com
Today, 12:02 AM
|
High-quality fungal reference genomes are essential for comparative, functional, and evolutionary studies, yet fungal genome features such as repeats, structural rearrangements, accessory chromosomes, and intron-rich genes can complicate genome assembly and the selection of cost-effective sequencing strategies. Here, we benchmark fungal genome assembly performance using simulated and empirical short- and long-read datasets to evaluate how sequencing depth, assembler choice, and genome characteristics influence contiguity, completeness, accuracy, and computational requirements. Using simulated reads from complete fungal genomes spanning diverse sizes and compositions, we evaluated short-read, long-read, hybrid, and polished long-read assemblies across sequencing depths from 10X to 100X. Key trends were validated using empirical sequencing data from 10 fungal isolates assembled with multiple strategies, including different Flye assembler parameter sensitivity and short-read polishing. Across datasets, long reads produced the largest improvements in contiguity, with most gains achieved at ~20-40X coverage and diminishing returns beyond moderate depth. Short-read polishing substantially improved base-level accuracy at relatively low cost, with ~10-20X coverage often sufficient to approach maximal error reduction. Hybrid assemblers showed strong algorithmic variability, with trade-offs between contiguity, error rates, and computational demand. Genome architecture also influenced outcomes, as larger and more feature-dense genomes benefited more from long-read data while GC content had limited impact. Overall, our results suggest that moderate long-read coverage (~30-40X) combined with modest short-read polishing (~10-20X), particularly using Flye plus Polypolish, provides a strong balance of contiguity, completeness, accuracy, and resource efficiency for generating high-quality fungal genome assemblies.
|
|
Scooped by
mhryu@live.com
June 8, 6:41 PM
|
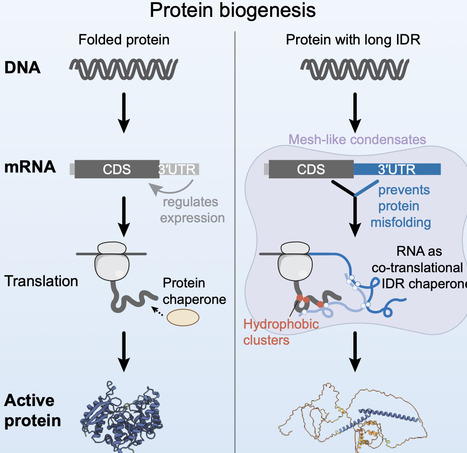
More than 2,700 human mRNA 3′ UTRs have hundreds of highly conserved nucleotides, but their biological roles are unclear. These mRNAs encode proteins strongly enriched for long intrinsically disordered regions (IDRs) with hydrophobic amino acid clusters. For MYC, UTX, and JMJD3, we show that their mRNA 3′ UTRs control protein activity. Rather than affecting protein abundance or localization, we find that the KDM6B 3′ UTR co-translationally changes the folding of JMJD3 protein. It promotes IDR-IDR interactions and suppresses folding between domains, suggesting that RNA has IDR chaperone activity that prevents interference between hydrophobic clusters in the IDR with folding of the structured domain. 3′ UTRs with chaperone activity are multivalent and mesh-like condensate-enriched, indicating the presence of localized folding environments for IDR-containing proteins. We show here that the protein sequence is insufficient for the biogenesis of fully active IDR-containing transcriptional regulators in cells, suggesting that mRNA 3′ UTRs control their activity by preventing co-translational misfolding.
|
|
Scooped by
mhryu@live.com
June 8, 6:18 PM
|
The recovery of metagenome-assembled genomes (MAGs) from shotgun metagenomic sequencing is rapidly expanding the availability of representative genomes. However, this practice may skew the representation of specific taxa in real-world datasets. This bias is attributed primarily to the known inefficiencies of sequence-by-synthesis platforms in amplifying GC-rich and AT-rich sequence fragments. Here, we recover 216 medium- and high-quality MAGs from an Australian wetland site. Notably, no MAGs were recovered for some dominant cyanobacterial and proteobacterial species known to be present. A new protocol involving read-based classification and alignment to the MAG dataset demonstrated the highly efficient recovery of low-GC organisms in the Actinobacteria and Bacteroidota phyla. Additionally, the recovery of lost taxonomic information was demonstrated through unmatched sample mapping. The findings suggest a bias towards the recovery of smaller, low-GC organisms in MAG recovery, potentially skewing the global representation of microbial diversity. Our pipeline is made publicly available as a tool to help researchers estimate taxonomic losses following MAG recovery efforts.
|
|
Scooped by
mhryu@live.com
June 8, 1:09 PM
|
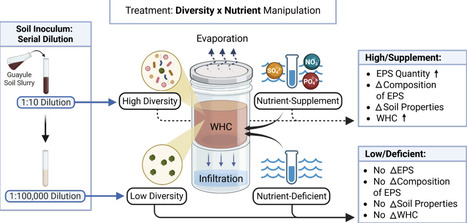
Soil microbial communities have a variety of mechanisms to deal with emerging drought stress. One well-documented mechanism is increased microbial production of extracellular polymeric substances which can potentially change the soil density and water holding capacity. Yet little is known about how microbial diversity influences the functional capacity of EPS formation and the resulting outcomes in water dynamics. To understand more about communal microbiome EPS production, we set up sterile mesocosms where we examined the effects of microbial diversity and nutrient input on these processes. To capture the microhydrology of the mesocosms, we measured water holding, infiltration, evaporation, and soil properties we believe microbes are altering. Our hypothesis stated that if diversity was artificially manipulated, then soil-water properties will be altered via production of EPS. We predicted that low diversity systems would have lower functional diversity, leading to less EPS production, moisture storage, and minimal changes from inert soil media. As predicted, we found that the high-diversity systems had a higher water retention and lower rates of water loss over time than low-diversity systems. This trend was magnified in the nutrient-supplemented treatment, suggesting that EPS production and subsequent water-holding traits are emergent features of the microbiome. Unexpectedly, we observed a correlation between the amount of water retained and the quantity of lipid EPS produced. This suggests that EPS composition, rather than quantity, is determinative of a biofilm's function. In conclusion, it appears that microbial diversity influences soil properties that are important to moisture retention within these systems. To date, the role that microbes and their diversity play in soil hydrology has been severely understudied, so this work aims to build ecological understandings of these systems. These findings are valuable, for if we learn how microbes manipulate soil moisture, we can apply these functions to advance sustainable agricultural practices and enhance ecosystem resilience to water scarcity in arid regions.
|
|
Scooped by
mhryu@live.com
June 8, 11:59 AM
|
The development of functional nanomaterials with controlled morphologies is essential for advancements in medicine, electronics and computing, energy, catalysis, and environmental applications. However, conventional synthesis methods often demand high energy input and pose significant environmental challenges. Urease-based biomineralization presents an efficient, eco-friendly alternative for nanomaterial production under mild conditions. In this study, we engineered E. coli to express a urease gene cluster from Sporosarcina pasteurii using CRAGE-Duet technology. The engineered strain successfully synthesized calcium carbonate and calcium phosphate crystals. Expanding the approach, we synthesized metal oxide nanoparticles, including hematite (Fe2O3), and nanocrystalline anatase titanium dioxide (TiO2). These nanomaterials were characterized by electron microscopy, demonstrating the potential of E. coli as a sustainable and versatile platform for green nanomaterial synthesis.
|
|
Scooped by
mhryu@live.com
June 8, 11:41 AM
|
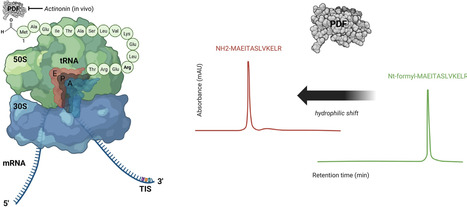
Accurate delineation of bacterial translation initiation sites (TISs) remains a major challenge, as conventional genome annotation and ribosome profiling (Ribo-seq) often lack the resolution to discriminate closely spaced start codons. To overcome these limitations, we developed TRAINSPOTTER (TRAnslation INitiation SPOTTER), a deformylation-assisted N-terminomics workflow that enables direct, proteome-wide detection of nascent N-termini indicative of active translation initiation. TRAINSPOTTER exploits the universal N-terminal formylation of initiator methionine in bacteria: in vitro enzymatic deformylation by peptide deformylase (PDF) generates a diagnostic hydrophilic shift, allowing selective isolation of previously formylated, initiation-derived peptides by COFRADIC-based chromatography. Optional in vivo PDF inhibition transiently enriches formylated N-termini, primarily enhancing detection sensitivity. Integration of pulse SILAC (Stable Isotope Labeling by Amino acids in Cell Culture) labeling confirmed that deformylation-shifted peptides represent newly synthesized N-termini, validating TRAINSPOTTER’s specificity for nascent translation products. Application to E. coli enabled precise mapping of >1000 TIS-indicative N-termini, including numerous alternative and near-cognate start sites, providing direct proteomic evidence for co-expressed N-terminal proteoforms. The method complements and refines Ribo-seq datasets, offering amino acid-level resolution for otherwise ambiguous initiation events. TRAINSPOTTER thus establishes a robust biochemical framework for proteome-wide identification of TISs and advances the experimental annotation of bacterial proteomes.
|
|
Scooped by
mhryu@live.com
June 8, 11:33 AM
|
Prokaryotic CRISPR–Cas systems rely on the Cas1–Cas2 protein complex to capture new DNA from mobile genetic elements (MGEs), to form immunological memory that defends against the MGEs. However, the mechanisms by which Cas1–Cas2 locates suitable DNA substrates inside cells remain unclear, limiting our understanding of how CRISPR–Cas immunity arises de novo. We directly visualized functional, DNA-bound Cas1–Cas2 complexes in bacteria, revealing the processes that license Cas1–Cas2 to capture DNA. Visible DNA-bound Cas1–Cas2 complexes formed only when replisomes are actively advancing, accumulating at post-replicative DNA gaps behind replication forks—structures arising during normal genome duplication, which are normally repaired by homologous recombination. Replication stress, which increases replicative DNA gap frequency, enhanced visible Cas1–Cas2 DNA binding. DNA capture by Cas1–Cas2 was strongly stimulated in cells lacking the RecFOR complex, which normally directs DNA gaps to repair. The RecBCD recombination initiator complex was essential for DNA capture by Cas1–Cas2 in these cells. The findings support a model in which naïve CRISPR–Cas adaptation is licensed by abundant replication-dependent DNA repair intermediates, prior to their repair by recombination. This identifies the mechanism co-ordinating Cas1–Cas2 with essential DNA replication and repair processes that all cells need, including when they are hijacked to replicate parasitic MGEs.
|


















zhao h,